
I am trying to perform a GO analysis of differentially expressed genes with GOseq, but I get a very poor fit of pwf by the nullp function.
A bit of context:
- I used bowtie to map the reads onto the transcriptome. Then salmon in alignment mode, tximport to import the results into R, and DESeq2 for the DGE analysis (FDR p< 0.001)
- I use all the genes obtained from salmon as assayed genes (which are actually all the genes in my transcriptome since I used the same data to assemble it)
- since my organism is not supported by GOseq, I created a vector with gene lengths from tximport count matrix with
lengthData <- rowMeans(count.matrix$length)which gives the mean across sample of transcript length in nucleotides
pwf <- nullp(DEgenes = go.seq.v,bias.data = lengthData) gives a weird fit
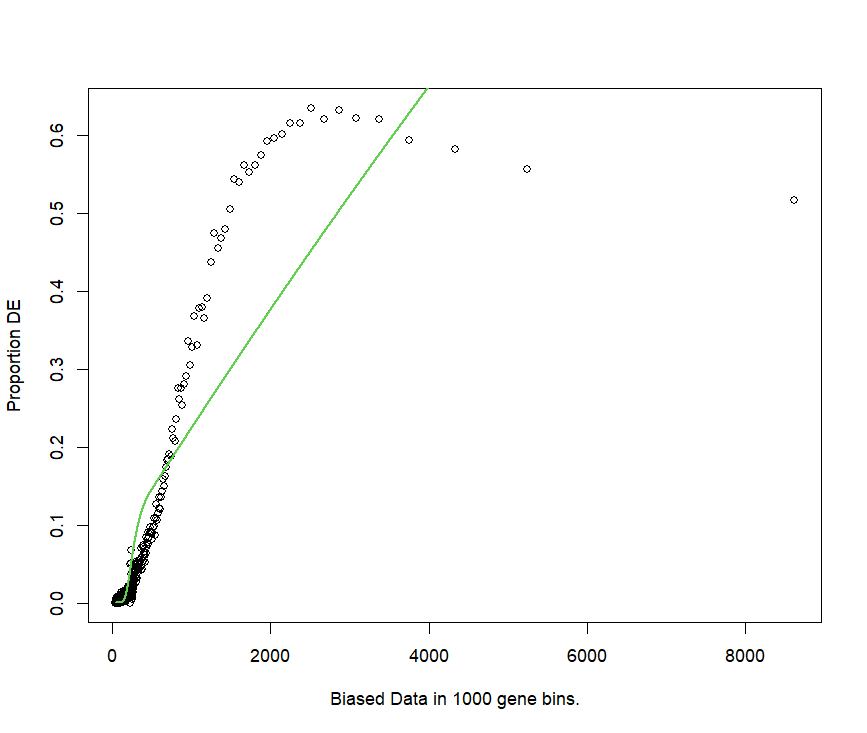
I tried to round the length values to the integer
lengthData.round <- as.integer(round(lengthData,-0))
pwf.r <- nullp(DEgenes = go.seq.v,bias.data = lengthData.round)
the fit is better, but the function shape is not what I expected at all.
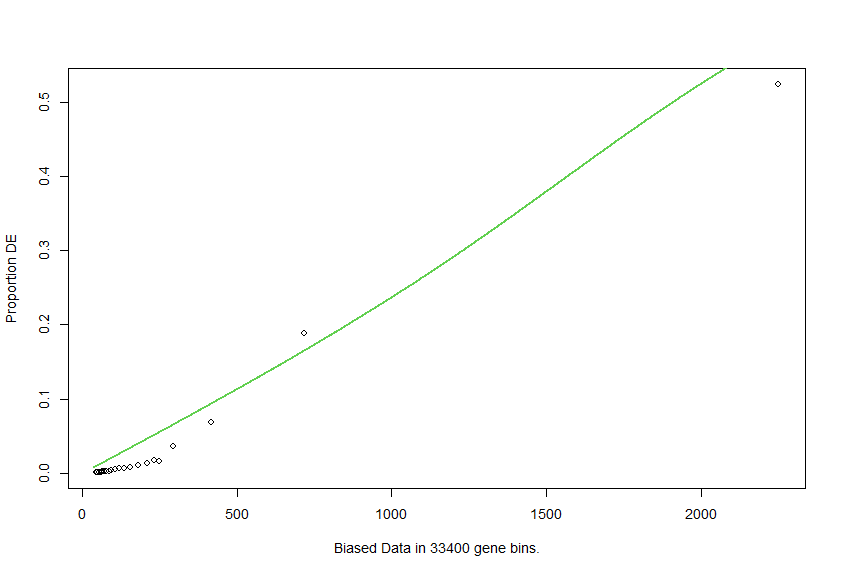
Hence my questions:
Is it correct to use float or integer for the length values? Why does it change the plot so much? Should I change something here
Could my DE genes not suffer from length bias? Maybe because DESeq2 is already accounting for it.
What shall I change to get a better fit? Can I change something in the model? Or should I work on the data?
Given that for whatever reason the pwf is not fitting the data well, would it make more sense not to account for gene length at all? Can I do this in GOseq or should I use another software like ClusterProfiler?
Thanks in advance for any input on the matter