Entering edit mode

@myprogramming2016-9741
Last seen 8.6 years ago
Hello,
I would like to use edgeR for differential Gene expression analysis.
I have read counts data on 50 individuals in three biological replicates.
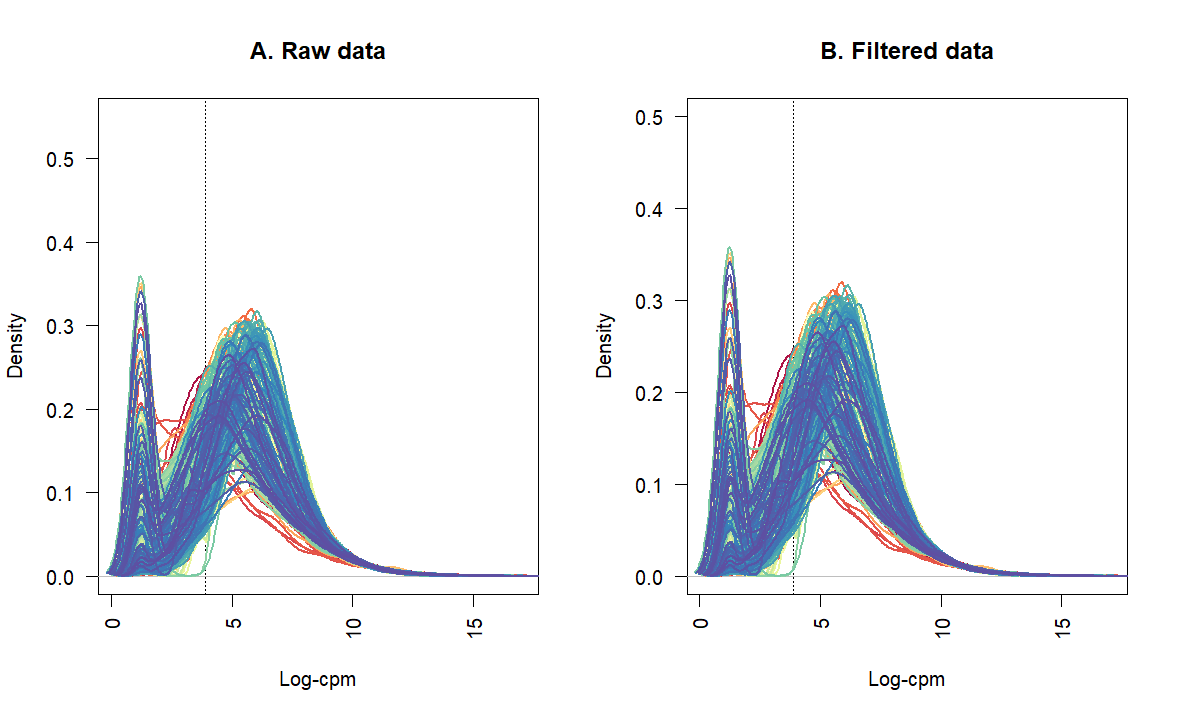
I would like to filter out lowly expressed genes. Is there a threshold to define express genes?
I was thinking to use CPM of >=2, and it should be in two of the three libraries.
How do I define R code?
Secondly, I would like to output TMM normalized counts to identify uniquely expressed genes in each of the individuals. How do I output the TMM counts in CPM?
Thanks in advance

Thanks for sharing an updated workflow. It is very helpful.
Secondly, I do not have a group (control Vs treated). I would like to identify differentially expressed genes in a particular individual compared with a mean of all the individuals (Intercept). I would like to repeat it for all the individuals.
I was wondering if there is any tutorial or an R script.
Thanks
Could you please reply to my post?
Thanks