
Hi Michael Love
I have RNASeq data for different layers of tissues that are stratified over one another in the same order. The library is SE unstranded data. I wish to perform DE and DEU RNAseq analysis to find out some novel patterns if present. (note: The library size for each tissue differs drastically from one another (even between replicates).
I obtained both genomic and transcriptomic bams using STAR and now I ran DESeq in the following way:
| Samples | group_time | group | Time |
|---|---|---|---|
| ALrep1_16daf | AL_16_daf | AL | 16_daf |
| ALrep2_16daf | AL_16_daf | AL | 16_daf |
| ALrep3_16daf | AL_16_daf | AL | 16_daf |
| CCrep1_16daf | CC_16_daf | CC | 16_daf |
| CCrep2_4daf | CC_4_daf | CC | 4_daf |
| NErep1_16daf | NE_16_daf | NE | 16_daf |
| NErep1_8daf | NE_8_daf | NE | 8_daf |
| NErep2_16daf | NE_16_daf | NE | 16_daf |
| NErep2_4daf | NE_4_daf | NE | 4_daf |
| NErep2_8daf | NE_8_daf | NE | 8_daf |
I wish to perform an all vs all comparison and though I know it's straight forward using contrast, I want to understand which is a valid approach:
Approach 1: Performing DESeq analysis on two samples at a time, say AL_16_daf.vs.NE_16_daf, which can be done by subsetting the above sample table for the replicates of these two samples, followed by DESeq analysis. This can be repeated for all pairwise comparisons that are desired.
Approach 2 The second approach could be the one described by Kevin here and by you here, where we take all the samples together, calculate the dispersion, and then using contrasts as described by Kevin can fish out results for each pairwise comparison.
On the same note, in DEXSeq, however, you can't perform such all vs all comparison using contrasts as mentioned here and here(also for DESeq) and therefore have to resort to something like I mentioned in Approach1 taking a pair at a time.
Having said that I have few queries:
So, to maintain uniformity in the analysis (both DESeq and DEXSeq), should I use Approach1 (wherein we estimate size factors and dispersion in a similar way for both DESeq and DEXseq, taking two tissues at a time and looping this over using, say, a FOR loop for all pairwise comparison). OR use Approach2 for DESeq2 and Approach1 for DEXSeq
What repercussions do you think one will have having used Approach1 over Approach2 and which one do you suggest. I did compare the list of DE genes generated by two approaches. All I could infer is the overlap between the DE genes obtained from both approaches is ~36% (based on two pairs that I compared ) and in some cases Approach 1 gives more DE genes and vice versa.

Hi Michael Love
So here are the pca plots for two design formulas:
plotPCA(vsd, intgroup = c("Sampletype"))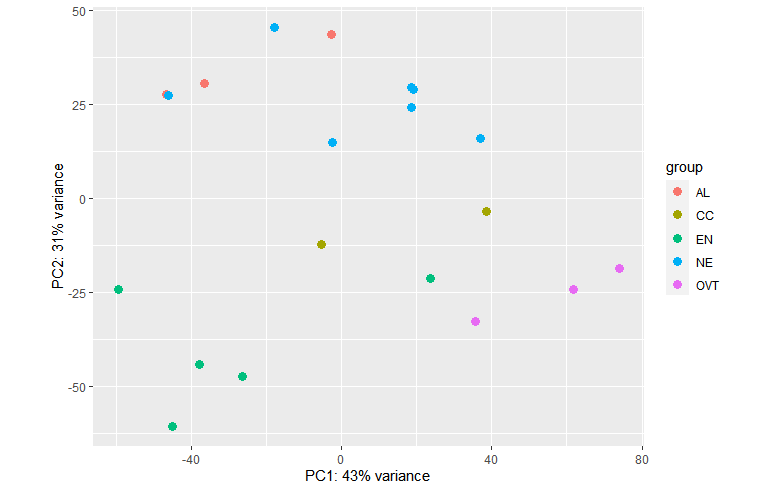 plotPCA(vsd, intgroup = c("Sampletype", "daf"))
plotPCA(vsd, intgroup = c("Sampletype", "daf"))
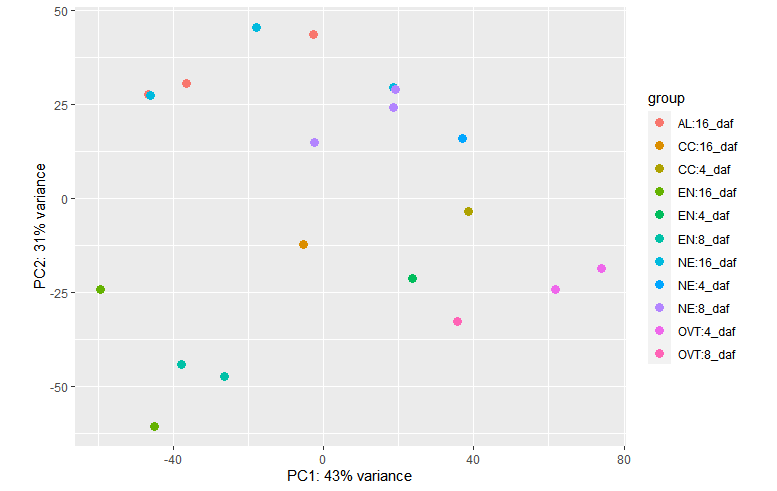 plotPCA(vsd, intgroup = c("daf"))
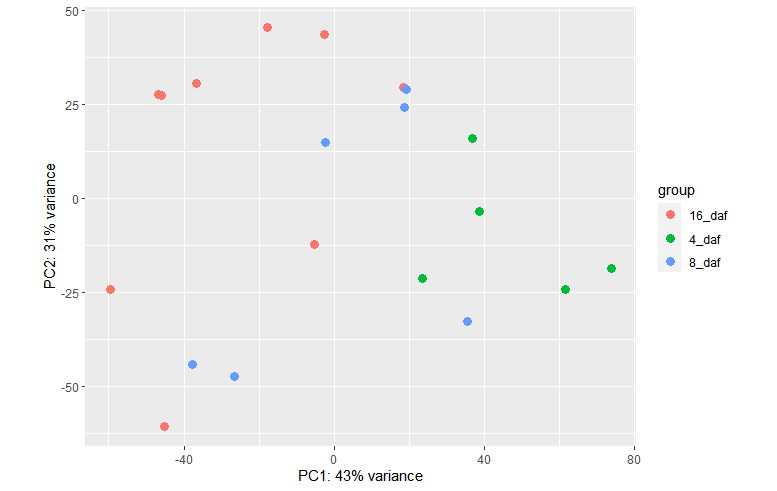
plotPCA(vsd, intgroup = c("daf"))
<h2>Hardcoding the sample type and days information</h2>Because I faced the error (mentioned below) when I tried introducing the interaction term Sampletype:daf, I separately ran DESeq2 for this condition to check if such grouping explained more variance than the above design formula:
plotPCA(vsd2, intgroup = c("group"))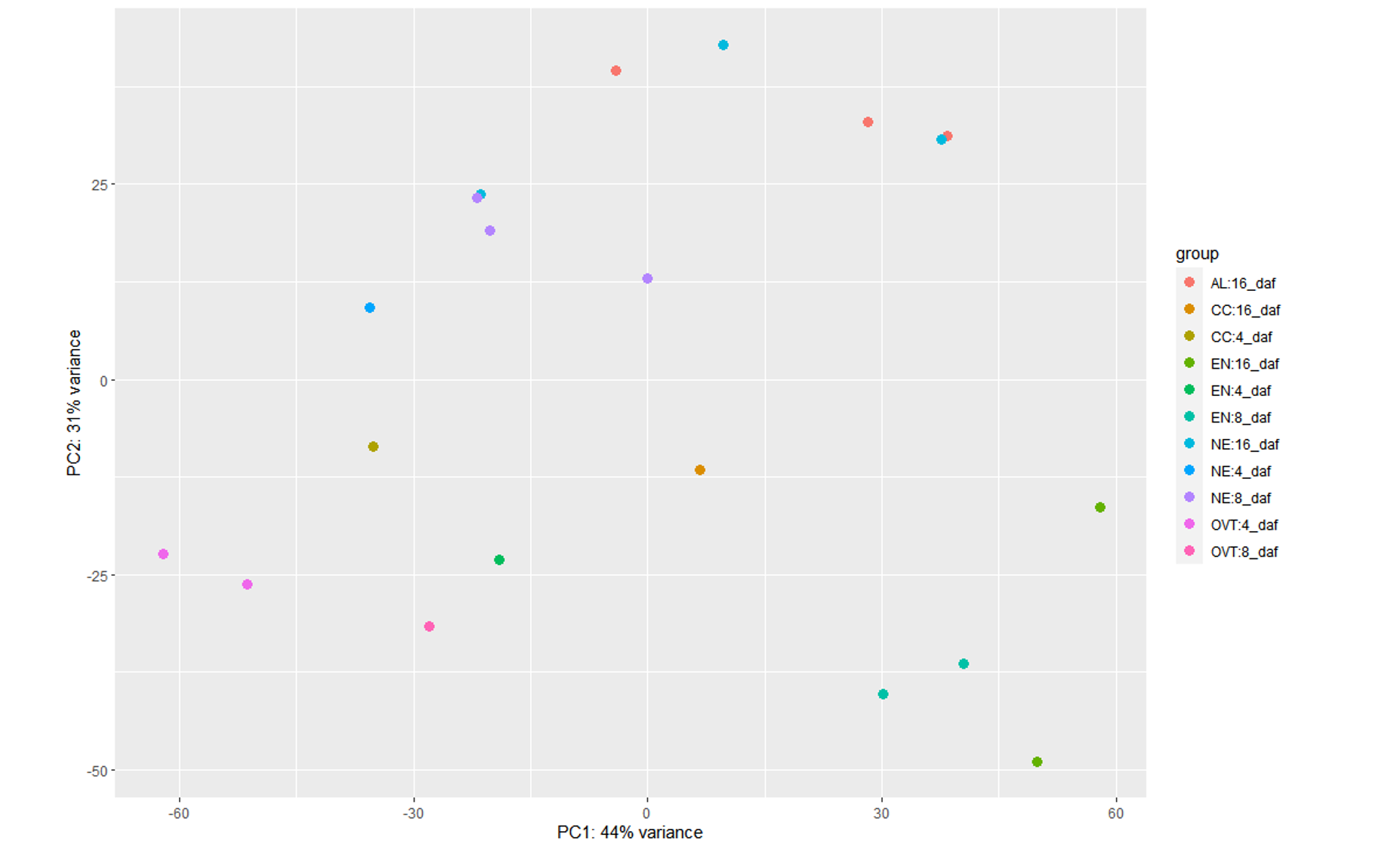
I would run these samples altogether I think.
Why do you say you can't use such models with DEXSeq?
"you can't perform such all vs all comparison using contrasts"
So Unlike DESeq wherein one can simply define contrast while using the
resultsfunction (you can do any sample vs any sample comparison), in DEXSeq, DESeqResults() function does not have that functionality (it compares all samples against the sample that comes first lexicographically i.e. all against one comparison)I see (I'm not a DEXSeq developer so I didn't realize the interface. I think you would just use pair-wise for DEXSeq, while the
contrastargument for DESeq2, unless there was a way to arrange it so it is an LRT within DEXSeq, but that's not obvious to me now.Hi Michael,
Can you elaborate on your comment in regards to using a PCA plot as guidance for a "all together" vs. "split by groups" approach? Thank you in advance.
See the FAQ in the vignette which elaborates on this, feel free to followup with questions as you need.