Entering edit mode

Sakthipriya
•
0
@sakthipriya-24094
Last seen 5.1 years ago
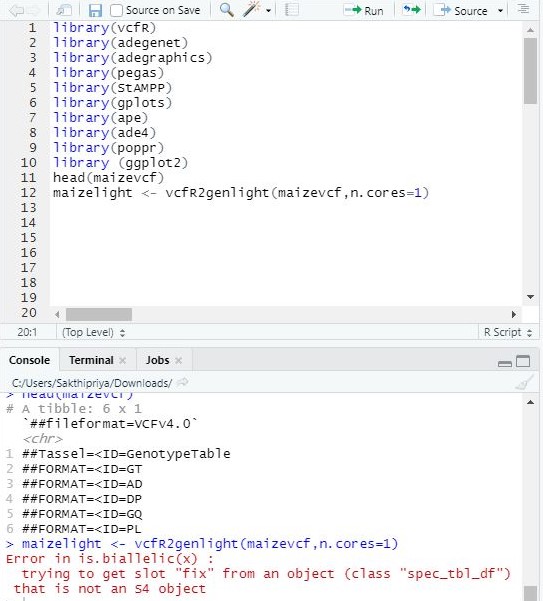
Hi,
I am trying to run the following using SNP data imported as vcf file, please see the image for details.
code run
maizelight <- vcfR2genlight(maizevcf,n.cores=1)
error obtained
Error in is.biallelic(x) :
trying to get slot "fix" from an object (class "spec_tbl_df") that is not an S4 object
anybody help to solve this error.
Thanks
-sakthi
vcfRis not a Bioconductor package; see https://cran.r-project.org/package=vcfR to contact the maintainer, perhaps through github.Thank you for the suggestion and help. will do that.
-Sakthi