Entering edit mode

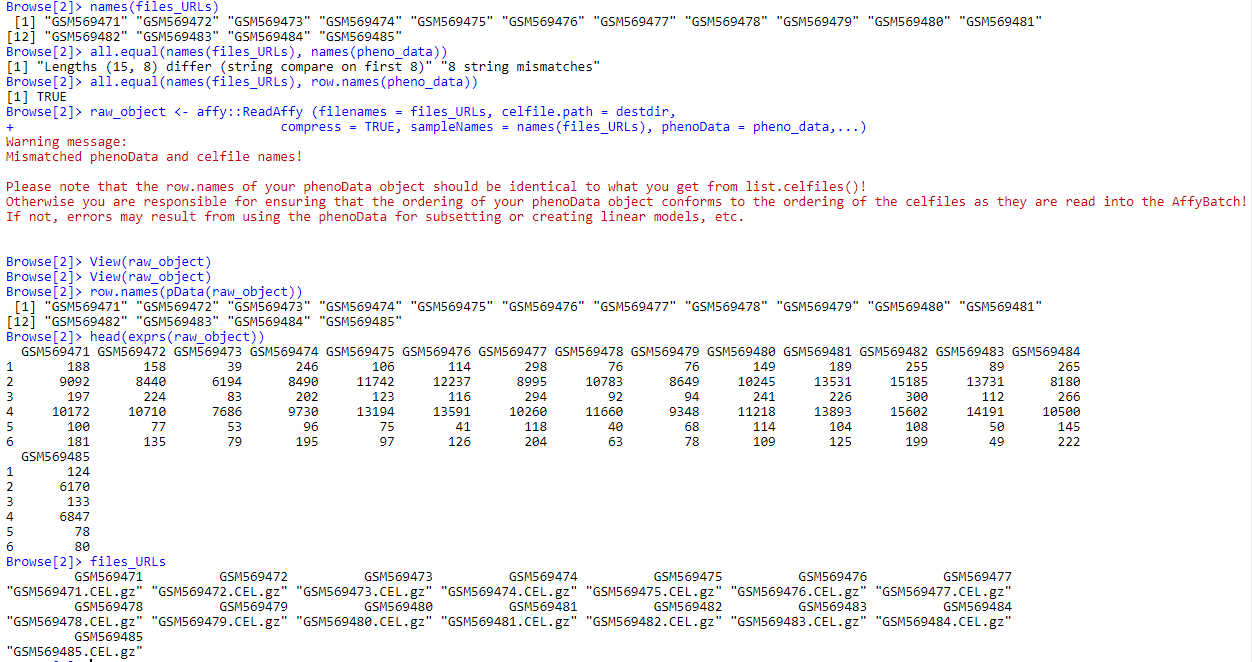
I am reading cel files using affy::readAffy functions. To that purpose, I used a named vector files_URLs (having used GEOquery to retrieve sample information), whose values are full names of the files, and names are the rownames I want to be used for my exprs and phenoData object (using argument sampleNames=names(files_URL)).
Operation is correctly performed (adequate files are read, with names being used for phenodata and exprs matrix as being defined by parameter sampleNames), however, i got following warning message (cf picture) when I perfom following instruction:
raw_object <- affy::ReadAffy (filenames = files_URLs, celfile.path = destdir, compress = TRUE, sampleNames = names(files_URLs), phenoData = pheno_data,...)
Warning message:
Mismatched phenoData and celfile names!
Please note that the row.names of your phenoData object should be identical to what you get from list.celfiles()!
Otherwise you are responsible for ensuring that the ordering of your phenoData object conforms to the ordering of the celfiles as they are read into the AffyBatch!
If not, errors may result from using the phenoData for subsetting or creating linear models, etc.
all.equal(names(files_URLs), row.names(pheno_data))
[1] TRUE
sessionInfo( )
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /softhpc/R/4.0.2/lib64/R/lib/libRblas.so
LAPACK: /softhpc/R/4.0.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8
[6] LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bmkanalysis_1.0.0

Thanks for your reply, but I don't want that the row.names of my phenoData match the files I read in in filenames, I want it to match, when provided, with argument sampleNames. Maybe a way of doing that properly would be to be able to provide a named vector for filenames argument: when you don't have names, function would perform as usual. But when you provide names to filenames, this would be used in place of argument
sampleNames, assuring both sampleNames of ÀffyBatch` object are the names given in named vector filenames, and the order is respected.This would besides remove an argument to function readAffy, replacing sampleNames by names, when avalaible, of filenames argument.
This isn't really an issue with the affy package, nor
ReadAffy. The issue is that you are creating aphenoDataobject that won't match other data in yourAffyBatch, and is getting rejected when it is tested for validity. In general I don't recommend making aphenoDataobject at the outset, which is why I added the warning that you saw in your original post, attempting to tell people exactly what the problem is.In other words, if you want to make your own
phenoDataobject and pass that into yourAffyBatch, then you have taken on the responsibility of ensuring it matches up correctly orvalidObjectwill tell you that your object isn't valid, as it should.Picking up where I left off,