
I have invented a dataset below (that mirrors my real data) with 3 factors - disease (sick/healthy), treatment (drug/saline/untreated), and cell line (#1 and #2). The cell lines are supposed to be my 2 replicates but plotMDS() of my data showed that they are quite different, so I made cell line a factor.
| Cell line | Disease | Treatment |
|---|---|---|
| #1 | Healthy | Drug |
| #1 | Healthy | Sal |
| #1 | Healthy | Unt |
| #1 | Sick | Drug |
| #1 | Sick | Sal |
| #1 | Sick | Unt |
| #2 | Healthy | Drug |
| #2 | Healthy | Sal |
| #2 | Healthy | Unt |
| #2 | Sick | Drug |
| #2 | Sick | Sal |
| #2 | Sick | Unt |
| ----------- | ------------- | ------------- |
I want to: 1) find the DEGs between Healthy and Sick in all cells, as well as in cell_line #1 and #2 separately 2) see if the drug (versus saline) has an effect on the sick cells. 3) confirm that Saline and Untreated cells do not differ (would it be best to do Saline vs Untreated or compare Drug vs Saline to Drug vs Untreated to see if they match?).
I made the following design matrices
design2 <- model.matrix(~Disease + Disease:Treatment + Cell_line)
design3 <- model.matrix(~Disease + Disease:Cell_line + Disease:Treatment)
colnames(design2)
#[1] "(Intercept)" "DiseaseSick" "Cell_line#2"
#[4] "DiseaseHealthy:TreatmentSal" "DiseaseSick:TreatmentSal" "DiseaseHealthy:TreatmentUnt"
#[7] "DiseaseSick:TreatmentUnt"
colnames(design3)
#[1] "(Intercept)" "DiseaseSick" "DiseaseHealthy:Cell_line#2"
#[4] "DiseaseSick:Cell_line#2" "DiseaseHealthy:TreatmentSal" "DiseaseSick:TreatmentSal"
#[7] "DiseaseHealthy:TreatmentUnt" "DiseaseSick:TreatmentUnt"
My questions today:
1) design2 gives me high dispersion values with plotBCV() with asymptote at 0.5, while design3 gives me much lower dispersion values with asymptote at 0.1 - Does this mean that design3 better approximates the relationships between the covariates, therefore is the better model for the data?
2) In both designs, is coef=2 considering Disease-Unt, Disease-Saline, and Disease-Drug as 6 sample replicates and comparing them to Healthy-Unt, Healthy-Saline, and Healthy-Drug? Is the model's adjustment for the treatment factor so complete that it can be ignored?? Would the DEGs generated for the 2 Disease-Unt vs 2 Healthy-Unt (no treatment effect included) samples be the same as the DEGs from coef=2 (6 Disease vs 6 Healthy samples)? Why does coef=2 in design2 and coef=2 in design3 give me different DEGs even though they compare the same samples?
3) I have a general question about interaction terms using DiseaseSick:Cell_line#2 as an example. From my understanding, specifying this coef gives me the differences (DEGs) between Cell_line #2 vs #1 in the Sick group. But what I want is the difference between Sick and Healthy in Cell_line#2 ! I tried inverting the term in the design matrix (see code below) to Cell_line:Disease but the interaction terms dont reverse! Am I missing something?
design3_reverse <- model.matrix(~Disease + Cell_line:Disease + Disease:Treatment)
colames(design3_reverse)
#[1] "(Intercept)" "DiseaseSick" "DiseaseHealthy:Cell_line#2"
#[4] "DiseaseSick:Cell_line#2" "DiseaseHealthy:TreatmentSal" "DiseaseSick:TreatmentSal"
#[7] "DiseaseHealthy:TreatmentUnt" "DiseaseSick:TreatmentUnt"
I hope someone can help explain how to extract what I need! I have not been able to find previous posts or examples in the UserGuide that are similar to my dataset. Session info is below.
sessionInfo( )
#R version 4.0.3 (2020-10-10)
#Platform: x86_64-w64-mingw32/x64 (64-bit)
#Running under: Windows 10 x64 (build 18363)
#Matrix products: default
#locale:
#[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252
#[3] LC_MONETARY=English_United States.1252 LC_NUMERIC=C
#[5] LC_TIME=English_United States.1252
#attached base packages:
#[1] parallel stats4 stats graphics grDevices utils datasets methods base
#other attached packages:
#[1] statmod_1.4.35 forcats_0.5.0 stringr_1.4.0 purrr_0.3.4
#[5] readr_1.4.0 tidyr_1.1.2 tibble_3.0.4 tidyverse_1.3.0
#[9] calibrate_1.7.7 MASS_7.3-53 knitr_1.30 org.Hs.eg.db_3.12.0
#[13] AnnotationDbi_1.52.0 IRanges_2.24.1 S4Vectors_0.28.1 Biobase_2.50.0
#[17] BiocGenerics_0.36.0 GOplot_1.0.2 gridExtra_2.3 ggdendro_0.1.22
#[21] goseq_1.42.0 geneLenDataBase_1.26.0 BiasedUrn_1.07 plotly_4.9.2.1
#[25] ggplot2_3.3.2 pheatmap_1.0.12 randomcoloR_1.1.0.1 RColorBrewer_1.1-2
#[29] magrittr_2.0.1 gplots_3.1.1 plyr_1.8.6 dplyr_1.0.2
#[33] caTools_1.18.0 edgeR_3.32.0 Glimma_2.0.0 limma_3.46.0
#loaded via a namespace (and not attached):
#[1] readxl_1.3.1 backports_1.2.0 BiocFileCache_1.14.0
#[4] lazyeval_0.2.2 splines_4.0.3 BiocParallel_1.24.1
#[7] GenomeInfoDb_1.26.2 digest_0.6.27 htmltools_0.5.0
#[10] GO.db_3.12.1 fansi_0.4.1 memoise_1.1.0
#[13] cluster_2.1.0 Biostrings_2.58.0 annotate_1.68.0
#[16] modelr_0.1.8 matrixStats_0.57.0 askpass_1.1
#[19] prettyunits_1.1.1 colorspace_2.0-0 blob_1.2.1
#[22] rvest_0.3.6 rappdirs_0.3.1 haven_2.3.1
#[25] xfun_0.19 crayon_1.3.4 RCurl_1.98-1.2
#[28] jsonlite_1.7.2 genefilter_1.72.0 survival_3.2-7
#[31] glue_1.4.2 gtable_0.3.0 zlibbioc_1.36.0
#[34] XVector_0.30.0 DelayedArray_0.16.0 V8_3.4.0
#[37] scales_1.1.1 DBI_1.1.0 Rcpp_1.0.5
#[40] viridisLite_0.3.0 xtable_1.8-4 progress_1.2.2
#[43] bit_4.0.4 htmlwidgets_1.5.3 httr_1.4.2
#[46] ellipsis_0.3.1 pkgconfig_2.0.3 XML_3.99-0.5
#[49] dbplyr_2.0.0 locfit_1.5-9.4 tidyselect_1.1.0
#[52] rlang_0.4.9 munsell_0.5.0 cellranger_1.1.0
#[55] tools_4.0.3 cli_2.2.0 generics_0.1.0
#[58] RSQLite_2.2.1 pacman_0.5.1 broom_0.7.3
#[61] evaluate_0.14 yaml_2.2.1 bit64_4.0.5
#[64] fs_1.5.0 nlme_3.1-149 xml2_1.3.2
#[67] biomaRt_2.46.0 compiler_4.0.3 rstudioapi_0.13
#[70] curl_4.3 reprex_0.3.0 geneplotter_1.68.0
#[73] stringi_1.5.3 GenomicFeatures_1.42.1 lattice_0.20-41
#[76] Matrix_1.2-18 vctrs_0.3.5 pillar_1.4.7
#[79] lifecycle_0.2.0 data.table_1.13.4 bitops_1.0-6
#[82] rtracklayer_1.49.5 GenomicRanges_1.42.0 R6_2.5.0
#[85] KernSmooth_2.23-17 gtools_3.8.2 assertthat_0.2.1
#[88] SummarizedExperiment_1.20.0 openssl_1.4.3 DESeq2_1.30.0
#[91] withr_2.3.0 GenomicAlignments_1.26.0 Rsamtools_2.6.0
# [94] GenomeInfoDbData_1.2.4 mgcv_1.8-33 hms_0.5.3
#[97] grid_4.0.3 rmarkdown_2.6 MatrixGenerics_1.2.0
#[100] Rtsne_0.15 lubridate_1.7.9.2
I thought I might be able to combine Disease and Treatment (Dis.Tr), and cell_line linearly to the model in the end, to make a single factorial design - model.matrix(~ 0 + Dis.Tr + cell_line). But the cell_line effect may not be the same across both genotypes (between Sick and Healthy), so I dont know if this is correct....
It would be nice to have a look at the MDS plot first. If a linear effect of the cell line is observed on the MDS plot, then it would be okay to use
design <- model.matrix(~ 0 + Dis.Tr + cell_line)Dear Yunshun, I am not sure how to determine a linear or non-linear effect from the MDS plot. Here is the info that I get from the MDS plot below - there are clear differences between Sick and Healthy samples (disease factor), as well as between #1 and #2 samples (cell-line factor), but there does not seem to be any effect of the drug (treatment factor highlighted by color). How do I check if the effect of each factor is linear or not?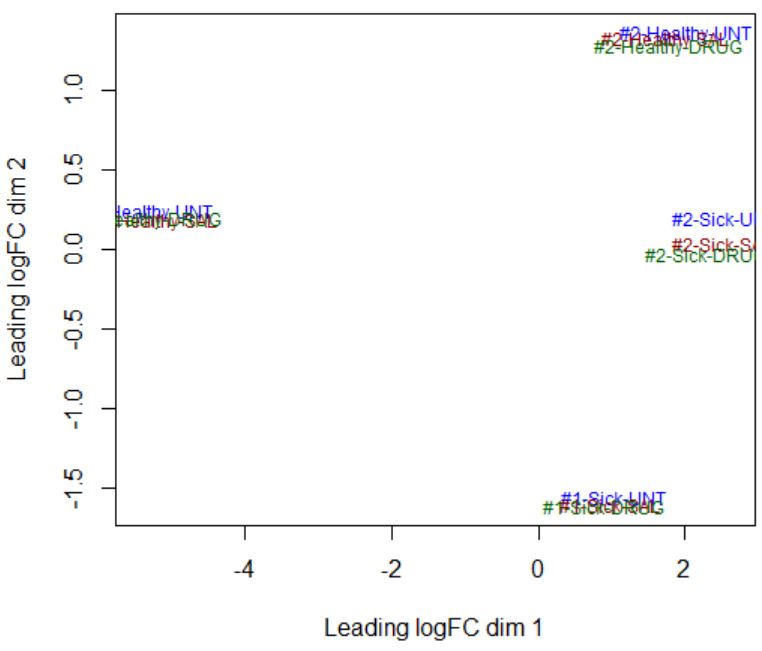
Take your MDS plot as an example, if the four clusters of samples are at the four corners (i.e., separated by cell_line in the dim1 and by Disease in dim 2), then I would say the cell_line effect is linear and it is okay to use
design <- model.matrix(~ 0 + Dis.Tr + cell_line). However, in your MDS plot the Healthy samples in cell line 1 are separated from the other groups in the first dimension. It isn't perfect but I think it should still be okay to use the additive model.ok I understand - Thank you! I will use this model for my dataset.
Would you be able to address my other questions from the original post? They are more general about the edgeR functions and understanding how they work and how to interpret the results they give. As you will see when you read the original post, my questions are specifically regarding plotBCV(), "coef" comparisons in glmQLFitTest(), and interaction terms in modelmatrix().