Entering edit mode

bio.erikson
•
0
@bioerikson-20673
Last seen 5.0 years ago
When I filter my count data with the code in the user guide, the FDR for all my genes drops to 1.0. But, if I don't filter or set the CPM cut off to ~0.2, then I start to get significant DE genes. I'm a bit confused by this behavior. What might be causing it?
My experiment is in Xenopus Laevis, where I have three conditions and four biological replicates. Each sample has a mean count of 8809887.
Running with Filtering
dge=DGEList(edge_data, group = edge_meta$group, genes = column_to_rownames(edge_anno, 'ID'))
keep <- rowSums(cpm(dge)>1) >= 2
dge <- dge[keep, , keep.lib.sizes=FALSE]
design=model.matrix(~Rep+Ploidy, data=edge_meta)
design
dge=calcNormFactors(dge)
dge=estimateDisp(dge,design)
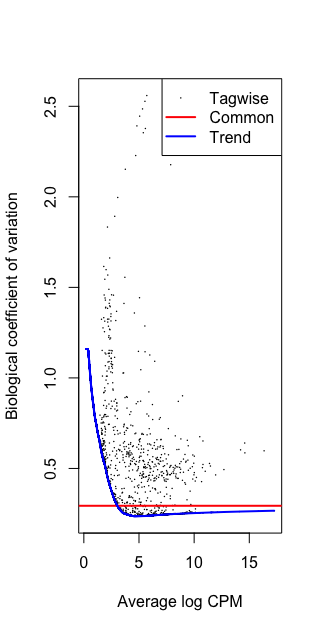
plotBCV(dge)
fit=glmQLFit(dge,design)
qlf=glmQLFTest(fit, coef=6)
topTags(qlf)
Coefficient: Ploidy3
logFC logCPM F PValue FDR
gene41545 2.0965805 2.448327 35.90115 4.746371e-05 0.9999619
gene49995 -0.7932641 4.473388 26.52267 1.941914e-04 0.9999619
gene15477 2.8440399 4.021164 26.31673 2.010534e-04 0.9999619
gene4260 1.1824923 4.668519 22.27339 4.144636e-04 0.9999619
gene50916 1.1398852 2.681193 19.93906 6.556942e-04 0.9999619
gene16190 1.6979906 5.217549 19.88785 6.625808e-04 0.9999619
gene16461 1.4116720 3.029733 17.98760 9.888933e-04 0.9999619
gene30142 2.2028513 1.304547 17.88017 1.012326e-03 0.9999619
gene9327 1.6603994 3.380958 17.84182 1.020847e-03 0.9999619
gene41018 0.5355154 4.970458 16.86752 1.267972e-03 0.9999619
Running w/o filtering
dge=DGEList(edge_data, group = edge_meta$group, genes = column_to_rownames(edge_anno, 'ID'))
#keep <- rowSums(cpm(dge)>1) >= 2
#dge <- dge[keep, , keep.lib.sizes=FALSE]
design=model.matrix(~Rep+Ploidy, data=edge_meta)
design
dge=calcNormFactors(dge)
dge=estimateDisp(dge,design)
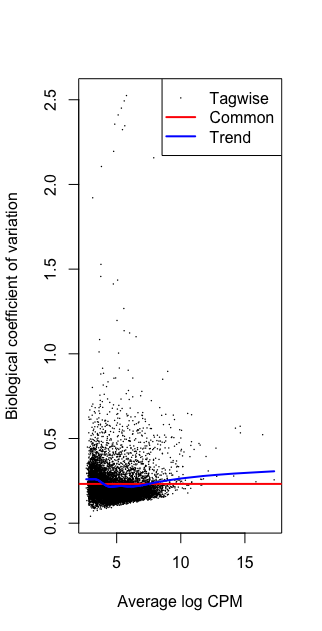
plotBCV(dge)
fit=glmQLFit(dge,design)
qlf=glmQLFTest(fit, coef=6)
topTags(qlf)
Coefficient: Ploidy3
logFC logCPM F PValue FDR
gene15477 2.8412637 4.012968 147.49207 6.236814e-34 3.189070e-29
gene16190 1.6964975 5.212556 109.25227 1.443137e-25 3.689595e-21
gene42380 1.7616811 4.875927 101.21749 8.312275e-24 1.416772e-19
gene18119 1.8589354 4.609218 100.35729 1.283117e-23 1.640241e-19
gene14278 1.2991257 6.502732 99.87230 1.638995e-23 1.676135e-19
gene45604 1.0203924 7.505369 72.36337 1.797921e-17 1.532218e-13
gene4166 0.9549254 7.469934 61.14997 5.305508e-15 3.875522e-11
gene46483 0.8449512 9.177001 60.20210 8.586239e-15 5.488002e-11
gene18489 2.2123988 2.705238 56.55142 5.489566e-14 3.118867e-10
gene13964 1.3560655 4.548504 55.43617 9.679432e-14 4.949384e-10
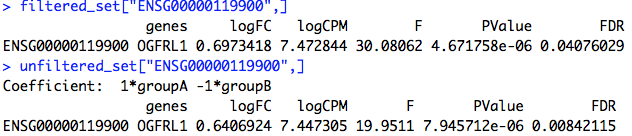
Gene CPM in each library


Cross-posted to Biostars: https://www.biostars.org/p/377604/