
Hello,
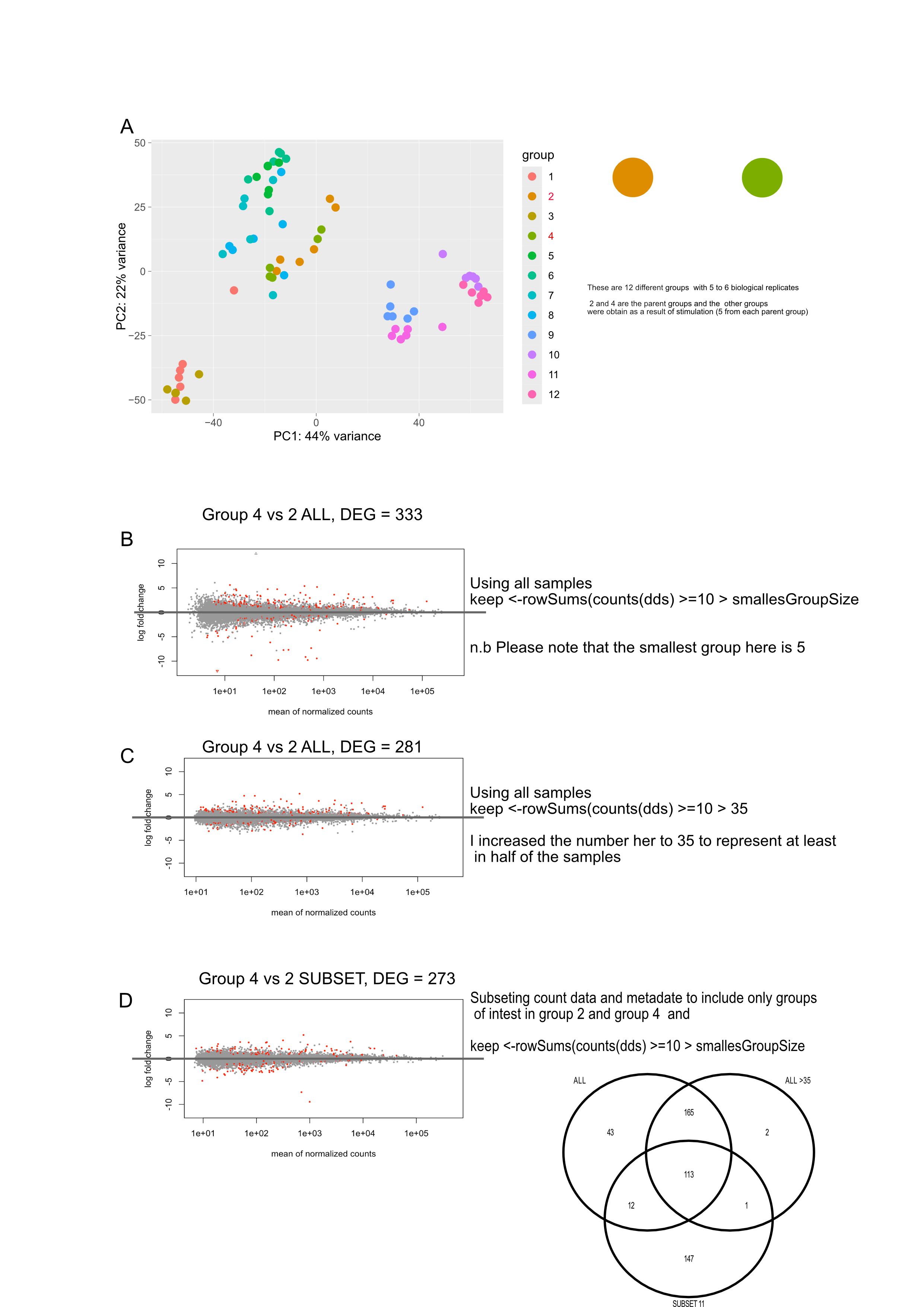
I've done some differential expression analysis with 3 main groups (3 or 4 biological replicates for each group, 11 total samples). When comparing any two of the groups, there are a number of genes that are statistically significant (in some cases, quite a lot) that have very high log2FoldChanges (~15-35) with high standard errors (~4). Upon further inspection, it seems what is happening with at least some of these genes is that only 1 or 2 replicates (out of 3 or 4) are expressing a gene with the other samples showing 0 expression/counts. Now these samples are from primary tissue, so I'm not surprised that there is quite a bit of variability. However, I'm not sure how to best deal with these genes, I understand that they might still be statistically significant but I'm not sure how to best deal with it.
MA-plot example here: https://imgur.com/a/bNOvVWY
So, I was wondering what the best way the handle this would be. The ways I have thought to do it are:
- Just leave it as is.
- Pre-filter the data to only include genes that have non-zero counts in at least 2 of the 11 total samples (or perhaps in at least 1 sample of each group)? - This should get rid of some of these cases?
- Set some sort of standard error filter (lfcSE), but I have no idea what exact value would be reasonable here or if this is reasonable at all.
- Should I use lfcShrink using apeglm? Currently these results are just using results(dds) with a lfcthreshold set. If I use lfcShrink, can I then use the shrunken log2 fold changes while still using the adjusted p values from results(dds)?
Any thoughts on these or any other suggestions would be greatly appreciated.
Thanks.
EDIT: It seems DESeq2 might not be removing any outliers based on the cookscutoff due to how my design and samples are set up (see comment below), however I am still not sure how to best deal with this. Apologies for not mentioning this.
Updated Sample Info:
Samples are from 3 different tissues (3 or 4 replicates per tissue) which are sometimes from the same mice (my design is ~mouse + tissue), however mostly there is only 1 or 2 samples from a mice.


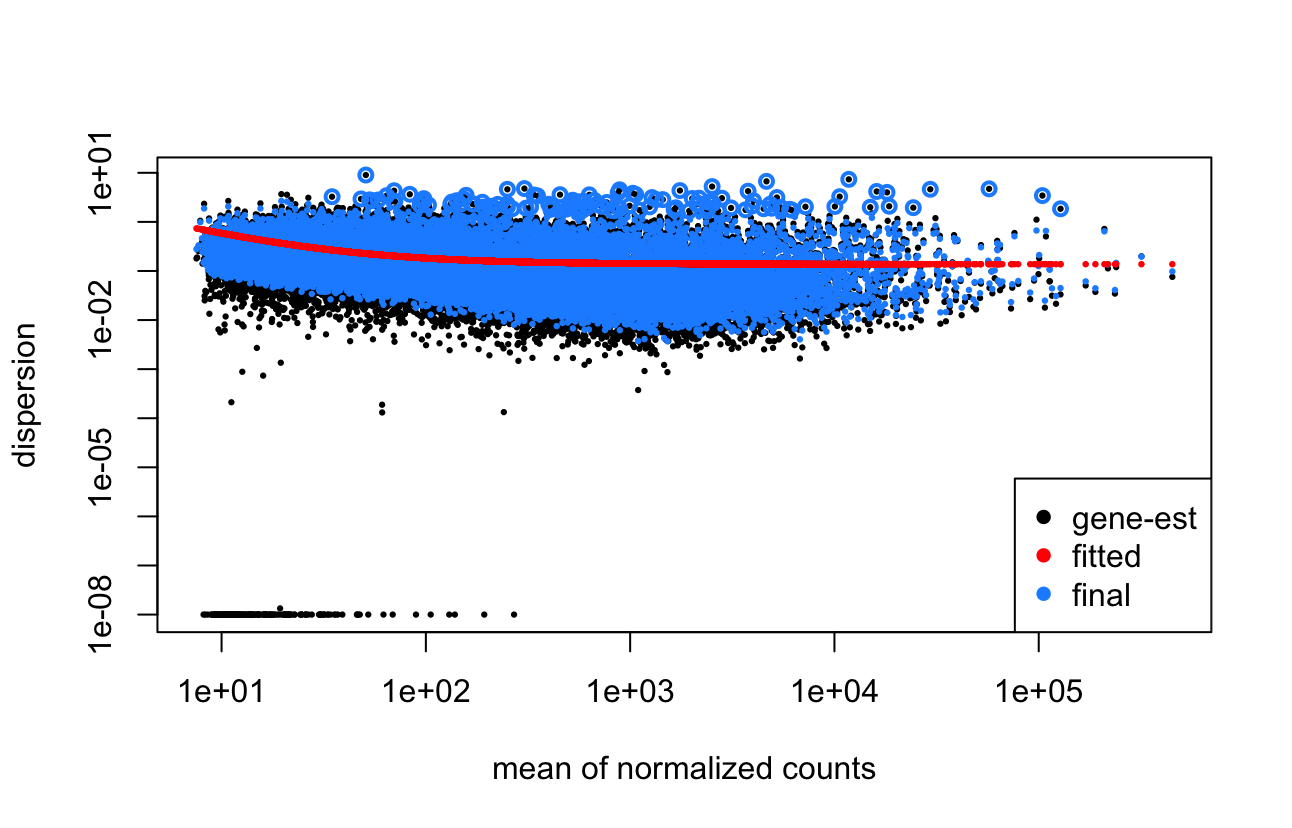 Seyram.
Seyram.
Thanks for your response
Looking into this more, I noticed that actually no genes are being filtered as outliers (i.e. based on cooksCutoff) and then I realised that this is due to my design (or at least I think it is). So essentially, my samples are from 3 different tissues which are sometimes from the same mice (my design is ~mouse + tissue), however mostly there is only 1 or 2 samples from a mice. And looking at the cookCutoff, it only works on samples that have at least 3 replicates and because of the mouse sample group, this isn't true for most of them. It seems like this might be the main issue as to why these genes are still being included. Sorry for not mentioning this.
However, I'm not sure what to do here. I could just remove mouse from the design (I can't really see any clear influence of it on a PCA plot) but I'm not sure this is ideal?
Thanks