
Hello everyone, hope you are doing well:)
I have already posted this question in Biostars, but didn't get an answer there, so I decided to post it here.
I have a question regarding making a heatmap to visualize significance AND presense of pathways across different conditions. I already did pathway enrichment analysis using gProfiler. I imported the results and manipulated them to make the names of the pathways as rownames, adjusted p values in rows and colnames for the conditions like so:
kegg_react_wp[1:3,] # showing only first 3 pathways
Condition1 Condition2 Condition3 Condition4 Condition5 Condition6
Pathway1 0.0003 0.3225 0.0540 0.0003 0.01 0.07
Pathway2 0.03 0.003225 0.0540 0.0003 0.01 0.07
Pathway3 0.703 0.3225 0.0540 0.0003 0.01 0.07
Because some pathways have an adjusted p value >0.05, I want to make these pathways as NA , which indicates that this pathway was NOT enriched in this condition. So I used the following code to do that as well as to -log10 the pathways with adjusted p value<0.05 so that most significant pathways have larger values:
Mooi_functie= function(x){
x = ifelse(x>0.05,NA,-log10(x))
return(x)
}
Now apply the function to my data
pathways_clean<-kegg_react_wp%>%
mutate_if(is.numeric,Mooi_functie) # this will make cells NA if their adjusted p value larger than 0.05
Now making the heatmap(using pheatmap or complexheatmap):
pheatmap(pathways_clean) # pheatmap package
Heatmap(pathways_clean)# complexheatmap package
I get the following error
Error in hclust(d, method = method) :
NA/NaN/Inf in foreign function call (arg 10) # from pheatmap
Error in hclust(get_dist(submat, distance), method = method) :
NA/NaN/Inf in foreign function call (arg 10) # from complexheatmap
A similar question has been posted here about the same error, but the purpose for that question, was different than mine. I want to KEEP NAs(unlike the already posted question) so that I can color them differently to indicate that these pathways are NOT present in this condition.
it seems that this error has to do with the clustering of the rows, so if I set cluster_rows=F, it works, but I want to cluster the rows and scale by row to see in which condition the significance is larger. I understand that there are some rows which are basiaclly all NAs except for one or two cells and this seems problematic for making the heatmaps.
I found in the internet a nice trick to add a column with any value , and it worked
pathways_clean$fake_column<-1
However, now It worked, but I am left with a fake column. it's not nice to have this column in the heatmap as you can see below.
the code I used to generate the heatmap:
pheatmap(pathways_clean,cluster_rows = T,na_col = "white",border_color = "white",
annotation_row =meta_kegg_wp_reac,cellwidth = 35,fontsize = 8,angle_col = 45,
scale = "row")
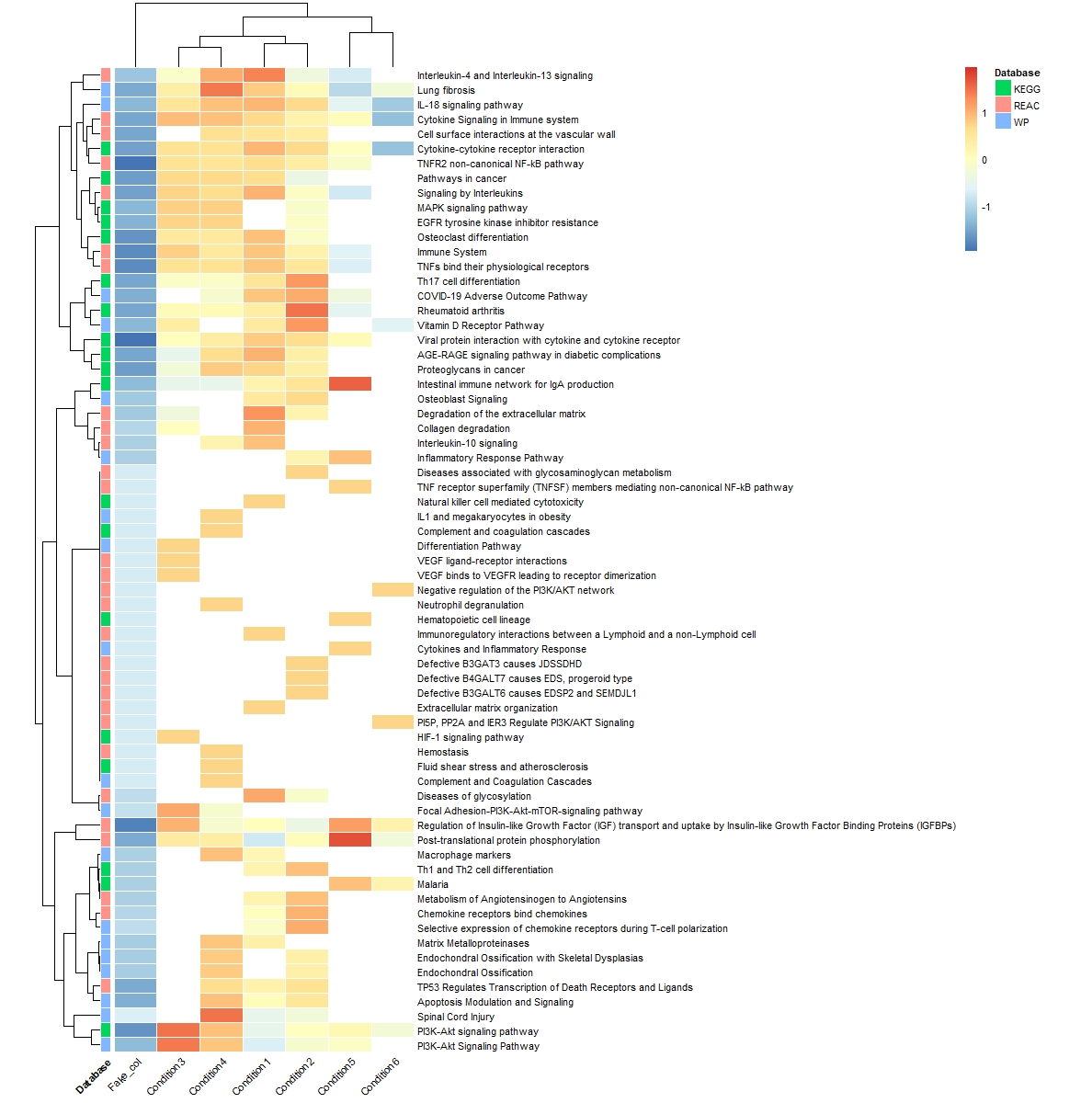
My questions are:
1) How to solve the issue of clustering with NAs without making a fake column? Or perhaps there is another way to visualize this without using NAs?(I tried replacing the NAs with zeros, but then the color scaling gets messy and cannot tell heads from tails).
2) Does it make sense to scale( scale="row") the -log10 of adjusted p values? Because I find it difficult to make sense of z-scores of -log10 of adjusted p values, which is what the legend in the heatmap represents.
3) Do you think that there might be a better way to visualize these results in my case?
Thank you very much in advance for your help!
**# My session info**
sessionInfo( )
R version 4.0.3 (2020-10-10)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 18363)
Matrix products: default
locale:
[1] LC_COLLATE=Dutch_Netherlands.1252 LC_CTYPE=Dutch_Netherlands.1252 LC_MONETARY=Dutch_Netherlands.1252
[4] LC_NUMERIC=C LC_TIME=Dutch_Netherlands.1252
attached base packages:
[1] stats4 grid parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] org.Hs.eg.db_3.11.4 AnnotationDbi_1.50.3 IRanges_2.22.2 S4Vectors_0.26.1 clusterProfiler_3.16.1
[6] treemap_2.4-2 gridBase_0.4-7 eulerr_6.1.0 ggupset_0.3.0 ComplexUpset_1.1.0
[11] wesanderson_0.3.6 viridis_0.5.1 viridisLite_0.3.0 networkD3_0.4 stringi_1.5.3
[16] tidyr_1.1.2 quantmod_0.4.18 TTR_0.24.2 xts_0.12.1 zoo_1.8-8
[21] UpSetR_1.4.0 nVennR_0.2.3 VennDiagram_1.6.20 futile.logger_1.4.3 gridExtra_2.3
[26] EnhancedVolcano_1.6.0 writexl_1.3.1 readr_1.4.0 scales_1.1.1 ggrepel_0.9.1
[31] esc_0.5.1 sjstats_0.18.1 sjmisc_2.8.6 sjlabelled_1.1.7 ggeffects_1.0.1
[36] strengejacke_0.5.0 sjPlot_2.8.7 pheatmap_1.0.12 cluster_2.1.1 digest_0.6.27
[41] circlize_0.4.12 RColorBrewer_1.1-2 ComplexHeatmap_2.4.3 ggpubr_0.4.0 ggplot2_3.3.3
[46] ggsci_2.9 limma_3.44.3 Biobase_2.48.0 BiocGenerics_0.34.0 data.table_1.13.6
[51] plyr_1.8.6 readxl_1.3.1 dplyr_1.0.4 readstata13_0.9.2
loaded via a namespace (and not attached):
[1] estimability_1.3 coda_0.19-4 bit64_4.0.5 knitr_1.31 multcomp_1.4-16
[6] generics_0.1.0 callr_3.5.1 cowplot_1.1.1 lambda.r_1.2.4 TH.data_1.0-10
[11] usethis_2.0.1 RSQLite_2.2.3 europepmc_0.4 listviewer_3.0.0 bit_4.0.4
[16] enrichplot_1.8.1 httpuv_1.5.5 xml2_1.3.2 assertthat_0.2.1 xfun_0.21
[21] hms_1.0.0 promises_1.2.0.1 evaluate_0.14 fansi_0.4.2 progress_1.2.2
[26] igraph_1.2.6 DBI_1.1.1 htmlwidgets_1.5.3 purrr_0.3.4 ellipsis_0.3.1
[31] selectr_0.4-2 backports_1.2.1 insight_0.13.1 grImport2_0.2-0 vctrs_0.3.6
[36] remotes_2.2.0 abind_1.4-5 cachem_1.0.3 withr_2.4.1 ggforce_0.3.2
[41] triebeard_0.3.0 emmeans_1.5.4 prettyunits_1.1.1 DOSE_3.14.0 crayon_1.4.1
[46] pkgconfig_2.0.3 labeling_0.4.2 tweenr_1.0.1 nlme_3.1-152 pkgload_1.1.0
[51] devtools_2.3.2 rlang_0.4.10 lifecycle_1.0.0 sandwich_3.0-0 downloader_0.4
[56] modelr_0.1.8 cellranger_1.1.0 rsvg_2.1 rprojroot_2.0.2 polyclip_1.10-0
[61] Matrix_1.2-18 urltools_1.7.3 carData_3.0-4 boot_1.3-27 base64enc_0.1-3
[66] ggridges_0.5.3 GlobalOptions_0.1.2 processx_3.4.5 png_0.1-7 rjson_0.2.20
[71] parameters_0.12.0 blob_1.2.1 shape_1.4.5 stringr_1.4.0 qvalue_2.20.0
[76] jpeg_0.1-8.1 rstatix_0.7.0 gridGraphics_0.5-1 ggsignif_0.6.1 memoise_2.0.0
[81] magrittr_2.0.1 compiler_4.0.3 scatterpie_0.1.5 clue_0.3-58 lme4_1.1-26
[86] snakecase_0.11.0 cli_2.3.1 patchwork_1.1.1 ps_1.5.0 formatR_1.7
[91] MASS_7.3-53.1 tidyselect_1.1.0 forcats_0.5.1 yaml_2.2.1 GOSemSim_2.14.2
[96] fastmatch_1.1-0 tools_4.0.3 rio_0.5.16 rstudioapi_0.13 foreign_0.8-81
[101] farver_2.0.3 ggraph_2.0.4 rvcheck_0.1.8 BiocManager_1.30.10 shiny_1.6.0
[106] Rcpp_1.0.6 car_3.0-10 broom_0.7.5 later_1.1.0.1 performance_0.7.0
[111] httr_1.4.2 effectsize_0.4.3 colorspace_2.0-0 polylabelr_0.2.0 rvest_0.3.6
[116] XML_3.99-0.5 fs_1.5.0 splines_4.0.3 statmod_1.4.35 graphlayouts_0.7.1
[121] ggplotify_0.0.5 sessioninfo_1.1.1 xtable_1.8-4 jsonlite_1.7.2 nloptr_1.2.2.2
[126] futile.options_1.0.1 tidygraph_1.2.0 testthat_3.0.2 R6_2.5.0 mime_0.10
[131] pillar_1.5.0 htmltools_0.5.1.1 glue_1.4.2 fastmap_1.1.0 minqa_1.2.4
[136] BiocParallel_1.22.0 codetools_0.2-16 fgsea_1.14.0 pkgbuild_1.2.0 mvtnorm_1.1-1
[141] utf8_1.1.4 lattice_0.20-41 tibble_3.0.6 curl_4.3 zip_2.1.1
[146] GO.db_3.11.4 openxlsx_4.2.3 survival_3.2-7 rmarkdown_2.7 desc_1.2.0
[151] munsell_0.5.0 DO.db_2.9 GetoptLong_1.0.5 haven_2.3.1 reshape2_1.4.4
[156] gtable_0.3.0 bayestestR_0.8.2

You can get an edge over the web with just visit the website and service that will also help you in find an online Quran Teacher website or you can Learners Quran Academy
Thank you sir for your comment. I thought of ignoring clustering by setting cluster.row=F. However, I am still faced with the problem of the colors. Because I have some -log10(fdr) of 30 and others around 5, the coloring of the heatmap looks not pretty. and you cannot really see contrasts. I tried making my own breaks, but it still looks very skewed towards high values without scaling. Any thoughts on how this can be done?
I would replace pvalue < 0.05 with -log10(0.05) instead of NA. NA makes those pvalues not taken into account, but those pvalues are known. They should not be ignored as if the other pvalues of the row are the only ones that allow a row to be compared another row. Whatever you choose, here how to your own clustering of rows:
Do your own scaling of -log10(pvalues). If current dynamic range is to big, compress it with one or two sqrt transformations.
If you kept the fake column for some reasons, remove it before display, then
Once you are OK with the transformation, you can try to play with breaks to associate the right value with a color. I usually don't take time to carry this out, as my goal of using pheatmap is only exploring relationships.
Of course everyone has to understand how to read a heatmap, ie link the color cells and the color scale, and on which values the trees were based. Are color encoded values are on an absolute scale or relative to each row or column? Are the trees of rows and/or columns based on the absolute values or relative values?
Think about the way you want to interpret the pvalues in your case...