
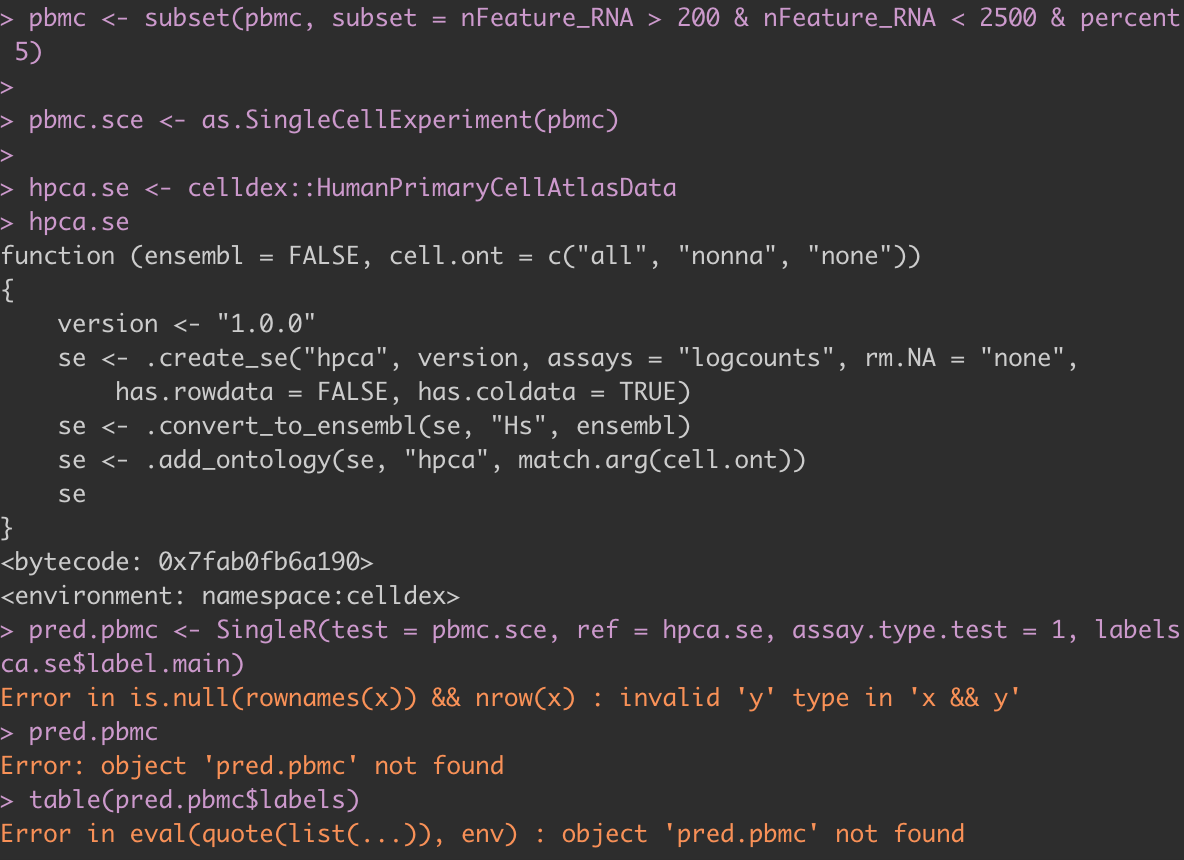
Hi, I have been having trouble using singleR. Every time I try to execute the singleR function I get this error and am uncertain what is causing this issue.
sessionInfo() R version 4.1.0 (2021-05-18) Platform: x86_64-apple-darwin17.0 (64-bit) Running under: macOS Catalina 10.15.7 (However, I get the same error from the same code when run on windows 10 through AWS) Matrix products: default BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
Random number generation: RNG: Mersenne-Twister Normal: Inversion Sample: Rounding
locale: 1 en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages: 1 parallel stats4 stats graphics grDevices utils datasets [8] methods base
other attached packages: 1 sctransform_0.3.2 SingleR_1.6.1 [3] celldex_1.2.0 scDblFinder_1.6.0 [5] scater_1.20.1 scuttle_1.2.0 [7] SingleCellExperiment_1.14.1 SummarizedExperiment_1.22.0 [9] Biobase_2.52.0 GenomicRanges_1.44.0 [11] GenomeInfoDb_1.28.1 IRanges_2.26.0 [13] S4Vectors_0.30.0 BiocGenerics_0.38.0 [15] MatrixGenerics_1.4.0 matrixStats_0.59.0 [17] ggplot2_3.3.5 patchwork_1.1.1 [19] SeuratObject_4.0.2 Seurat_4.0.3 [21] dplyr_1.0.7
loaded via a namespace (and not attached): 1 utf8_1.2.1 reticulate_1.20 [3] tidyselect_1.1.1 RSQLite_2.2.7 [5] AnnotationDbi_1.54.1 htmlwidgets_1.5.3 [7] grid_4.1.0 BiocParallel_1.26.1 [9] Rtsne_0.15 munsell_0.5.0 [11] ScaledMatrix_1.0.0 codetools_0.2-18 [13] ica_1.0-2 statmod_1.4.36 [15] scran_1.20.1 xgboost_1.4.1.1 [17] future_1.21.0 miniUI_0.1.1.1 [19] withr_2.4.2 argparse_2.0.3 [21] colorspace_2.0-2 filelock_1.0.2 [23] ROCR_1.0-11 tensor_1.5 [25] listenv_0.8.0 labeling_0.4.2 [27] GenomeInfoDbData_1.2.6 polyclip_1.10-0 [29] farver_2.1.0 bit64_4.0.5 [31] TH.data_1.0-10 coda_0.19-4 [33] parallelly_1.26.1 vctrs_0.3.8 [35] generics_0.1.0 lambda.r_1.2.4 [37] BiocFileCache_2.0.0 fastcluster_1.2.3 [39] doParallel_1.0.16 R6_2.5.0 [41] ggbeeswarm_0.6.0 rsvd_1.0.5 [43] locfit_1.5-9.4 reshape_0.8.8 [45] bitops_1.0-7 spatstat.utils_2.2-0 [47] cachem_1.0.5 DelayedArray_0.18.0 [49] assertthat_0.2.1 promises_1.2.0.1 [51] scales_1.1.1 multcomp_1.4-17 [53] beeswarm_0.4.0 gtable_0.3.0 [55] beachmat_2.8.0 globals_0.14.0 [57] goftest_1.2-2 sandwich_3.0-1 [59] rlang_0.4.11 splines_4.1.0 [61] lazyeval_0.2.2 spatstat.geom_2.2-0 [63] BiocManager_1.30.16 yaml_2.2.1 [65] reshape2_1.4.4 abind_1.4-5 [67] httpuv_1.6.1 tools_4.1.0 [69] gplots_3.1.1 ellipsis_0.3.2 [71] spatstat.core_2.2-0 RColorBrewer_1.1-2 [73] phyclust_0.1-30 ggridges_0.5.3 [75] Rcpp_1.0.7 plyr_1.8.6 [77] sparseMatrixStats_1.4.0 zlibbioc_1.38.0 [79] purrr_0.3.4 RCurl_1.98-1.3 [81] rpart_4.1-15 deldir_0.2-10 [83] pbapply_1.4-3 viridis_0.6.1 [85] cowplot_1.1.1 zoo_1.8-9 [87] ggrepel_0.9.1 cluster_2.1.2 [89] magrittr_2.0.1 futile.options_1.0.1 [91] data.table_1.14.0 scattermore_0.7 [93] lmtest_0.9-38 RANN_2.6.1 [95] mvtnorm_1.1-2 fitdistrplus_1.1-5 [97] mime_0.11 xtable_1.8-4 [99] gridExtra_2.3 compiler_4.1.0 [101] tibble_3.1.2 KernSmooth_2.23-20 [103] crayon_1.4.1 htmltools_0.5.1.1 [105] mgcv_1.8-36 later_1.2.0 [107] libcoin_1.0-8 tidyr_1.1.3 [109] DBI_1.1.1 formatR_1.11 [111] ExperimentHub_2.0.0 dbplyr_2.1.1 [113] MASS_7.3-54 rappdirs_0.3.3 [115] Matrix_1.3-4 metapod_1.0.0 [117] igraph_1.2.6 pkgconfig_2.0.3 [119] coin_1.4-1 plotly_4.9.4.1 [121] spatstat.sparse_2.0-0 foreach_1.5.1 [123] vipor_0.4.5 dqrng_0.3.0 [125] XVector_0.32.0 stringr_1.4.0 [127] digest_0.6.27 RcppAnnoy_0.0.18 [129] spatstat.data_2.1-0 Biostrings_2.60.1 [131] leiden_0.3.8 uwot_0.1.10 [133] edgeR_3.34.0 DelayedMatrixStats_1.14.0 [135] curl_4.3.2 gtools_3.9.2 [137] modeltools_0.2-23 shiny_1.6.0 [139] lifecycle_1.0.0 nlme_3.1-152 [141] jsonlite_1.7.2 BiocNeighbors_1.10.0 [143] futile.logger_1.4.3 viridisLite_0.4.0 [145] limma_3.48.1 fansi_0.5.0 [147] pillar_1.6.1 lattice_0.20-44 [149] KEGGREST_1.32.0 fastmap_1.1.0 [151] httr_1.4.2 survival_3.2-11 [153] interactiveDisplayBase_1.30.0 glue_1.4.2 [155] iterators_1.0.13 png_0.1-7 [157] bluster_1.2.1 BiocVersion_3.13.1 [159] bit_4.0.4 stringi_1.6.2 [161] blob_1.2.1 BiocSingular_1.8.1 [163] AnnotationHub_3.0.1 caTools_1.18.2 [165] memoise_2.0.0 ape_5.5 [167] irlba_2.3.3 future.apply_1.7.0

Hi, I met the same problem, have you solved it?