
Hello,
I am performing DTU analysis following "Swimming downstream: statistical analysis of differential transcript usage following Salmon quantification" publication workflow with own data (Link). My data include patient sample data, 21 samples on each 3 groups. I have used StringTie produced .ctab files from each sample as input for the workflow. First rows showed below:
>t_id chr strand start end t_name num_exons length gene_id gene_name cov FPKM
1 chr1 + 11874 14409 rna0 3 1652 DDX11L1 DDX11L1 0.286368 0.090211
2 chr1 - 14362 29370 rna1 11 1769 WASH7P WASH7P 5.673654`
I used tximport to download the data into R and to create txi object with transcript-to-gene-mapping object. Then I followed the workflow where Tx2gene was used as transcript-to-gene-mapping object. In this modification, t_name correspond to TXNAME and gene_name to GENEID. Tx2gene:
>A tibble: 76,103 × 3
t_name gene_name ntx
<chr> <chr> <table>
1 rna0 DDX11L1 1
2 rna1 WASH7P 1
3 rna3 MIR6859-1 3
4 rna2 MIR6859-1 3
5 rna4 MIR6859-1 3
6 rna5 MIR1302-2 2
Now, my problem is that for some reason DRIMSeq object inform me to have only 1 gene and 63 samples. The used code is below:
#DEXSeq_stringtie.csv file include the path to the files
DEXSeq_stringtie <- read_csv("DEXSeq_stringtie.csv")
path<-DEXSeq_stringtie$Path
tx2gene <- tmp[, c("t_name", "gene_name")]
txi <- tximport(path, type = "stringtie", tx2gene = tx2gene, txOut=TRUE, countsFromAbundance="scaledTPM")
#This was modified from both workflows
*reading in files with read_tsv
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 *
cts <- txi$counts
cts <- cts[rowSums(cts) > 0,]
#Checking data
all(rownames(cts) %in% tx2gene$t_name)
[1] TRUE
tx2gene <- tx2gene[match(rownames(cts),tx2gene$t_name),]
all(rownames(cts) == tx2gene$t_name)
[1] TRUE
#Adding metadata
samps<-Rmetadata_DRIMSeq
#Variables to factors
samps$group <- factor(samps$group)
samps$patient<- factor(samps$patient)
samps$diabetes<-factor(samps$diabetes)
samps<-as.data.frame(samps)
#DRIMSeq object
colnames(cts)<-samps$sample_id
counts <- data.frame(gene_id=tx2gene$gene_name, feature_id=tx2gene$t_name, cts)
d <- dmDSdata(counts=counts, samples=samps)
d
>An object of class dmDSdata
**with 1 genes and 63 samples** * data accessors: counts(), samples()
Would you think any reason why the object is giving me only 1 gene with the object? I appreciate your help!
sessionInfo(
R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /usr/local/apps/linux-centos7-x86_64/gcc-4.8.5/r/4.0.0-rmu2ifmqdxpwpvbr62zkux2ex77isony/rlib/R/lib/libRblas.so
LAPACK: /usr/local/apps/linux-centos7-x86_64/gcc-4.8.5/r/4.0.0-rmu2ifmqdxpwpvbr62zkux2ex77isony/rlib/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8
[2] LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8
[6] LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8
[8] LC_NAME=C
[9] LC_ADDRESS=C
[10] LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8
[12] LC_IDENTIFICATION=C
attached base packages:
[1] stats4 parallel stats graphics
[5] grDevices utils datasets methods
[9] base
other attached packages:
[1] tximport_1.18.0
[2] stageR_1.12.0
[3] DEXSeq_1.36.0
[4] RColorBrewer_1.1-2
[5] AnnotationDbi_1.52.0
[6] DESeq2_1.30.1
[7] SummarizedExperiment_1.20.0
[8] GenomicRanges_1.42.0
[9] GenomeInfoDb_1.26.7
[10] IRanges_2.24.1
[11] S4Vectors_0.28.1
[12] MatrixGenerics_1.2.1
[13] matrixStats_0.60.0
[14] Biobase_2.50.0
[15] BiocGenerics_0.36.1
[16] BiocParallel_1.24.1
[17] DRIMSeq_1.18.0
[18] edgeR_3.32.1
[19] limma_3.46.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7
[2] bit64_4.0.5
[3] progress_1.2.2
[4] httr_1.4.2
[5] tools_4.0.0
[6] utf8_1.2.2
[7] R6_2.5.0
[8] DBI_1.1.1
[9] colorspace_2.0-2
[10] tidyselect_1.1.1
[11] gridExtra_2.3
[12] prettyunits_1.1.1
[13] bit_4.0.4
[14] curl_4.3.2
[15] compiler_4.0.0
[16] cli_3.0.1
[17] xml2_1.3.2
[18] DelayedArray_0.16.3
[19] rtracklayer_1.50.0
[20] scales_1.1.1
[21] genefilter_1.72.1
[22] askpass_1.1
[23] rappdirs_0.3.3
[24] Rsamtools_2.6.0
[25] stringr_1.4.0
[26] DOSE_3.16.0
[27] XVector_0.30.0
[28] pkgconfig_2.0.3
[29] dbplyr_2.1.1
[30] fastmap_1.1.0
[31] rlang_0.4.11
[32] rstudioapi_0.13
[33] RSQLite_2.2.7
[34] generics_0.1.0
[35] hwriter_1.3.2
[36] GOSemSim_2.16.1
[37] dplyr_1.0.7
[38] RCurl_1.98-1.3
[39] magrittr_2.0.1
[40] GO.db_3.12.1
[41] GenomeInfoDbData_1.2.4
[42] Matrix_1.3-4
[43] Rcpp_1.0.7
[44] munsell_0.5.0
[45] fansi_0.5.0
[46] lifecycle_1.0.0
[47] stringi_1.7.3
[48] yaml_2.2.1
[49] zlibbioc_1.36.0
[50] plyr_1.8.6
[51] qvalue_2.22.0
[52] BiocFileCache_1.14.0
[53] grid_4.0.0
[54] blob_1.2.2
[55] DO.db_2.9
[56] crayon_1.4.1
[57] lattice_0.20-44
[58] Biostrings_2.58.0
[59] splines_4.0.0
[60] GenomicFeatures_1.42.3
[61] annotate_1.68.0
[62] hms_1.1.0
[63] locfit_1.5-9.4
[64] pillar_1.6.2
[65] fgsea_1.16.0
[66] geneplotter_1.68.0
[67] reshape2_1.4.4
[68] biomaRt_2.46.3
[69] fastmatch_1.1-3
[70] XML_3.99-0.6
[71] glue_1.4.2
[72] data.table_1.14.0
[73] vctrs_0.3.8
[74] gtable_0.3.0
[75] openssl_1.4.4
[76] purrr_0.3.4
[77] assertthat_0.2.1
[78] cachem_1.0.5
[79] ggplot2_3.3.5
[80] xtable_1.8-4
[81] survival_3.2-11
[82] tibble_3.1.3
[83] GenomicAlignments_1.26.0
[84] memoise_2.0.0
[85] statmod_1.4.36
[86] ellipsis_0.3.2)

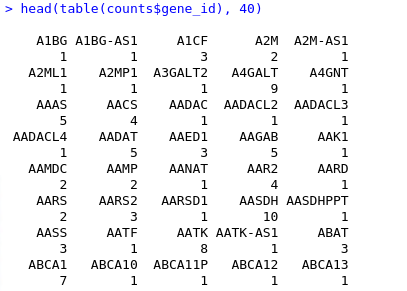
Ok, I reconstructed my initial input to tximport and I think dmDSdata object is as it should.
Prepping sample for tximport
..................
An object of class dmDSdata with 26895 genes and 63 samples