Entering edit mode

Hi Everyone!!
I am trying out a Package Gviz to make chromosome-wise coverage plot for a genome sequencing experiment. I was planning to plot per-chromosome-multi-sample coverage plot as shown below.
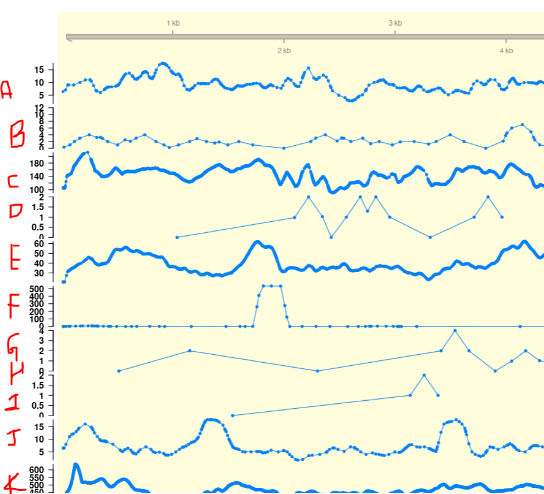
For that I have written a function that does that but now I wish to loop over that function and iteratively save the pdf. But I am unable to achieve that.
## Function
plotGenCov <- function(df, list.files, chr, labels=FALSE,regex="NEB.*",...){
options(ucscChromosomeNames=FALSE)
dtrack.all <- lapply(1:length(list.files), function(x){
if(isFALSE(labels)){
name <- gsub(regex,"",basename(list.files[x]))
DataTrack(range = list.files[x], type = "l", window = -1, name = name)
} else {
name <- labels[x]
DataTrack(range = list.files[x], type = "l", window = -1, name = name)
}
})
t <- GRanges(df)
gtrack<- GenomeAxisTrack(t[seqnames(t) == chr])
plotTracks(trackList = c(list(gtrack),dtrack.all),
chromosome = chr,
from=as.data.frame(t[seqnames(t) == chr])$start,
to=as.data.frame(t[seqnames(t) == chr])$end, cex.axis = 1, cex.title = 1,
col.title = "black", col.axis = "black",type = c("a", "p", "confint"),
...)
}
## Trying to loop over the function above
for (i in length(df$chrom)){
pdf(paste0(df$chrom[i],".pdf"),width = 15, height = 13)
grid::grid.newpage()
grid::grid.text(df$chrom[i],x = (0.5), y = (0.6),gp = gpar(fontsize = 18, fontface = "bold", fontfamily="Times"))
plotGenCov(df=df,chr=df$chrom[i],list.files = list.files,regex = "_NEB.*",
margin = 30, fontsize=10,background.panel = "#FFFEDB" , background.title="white",
labels = c("Sample a","Sample b","Sample c","Sample d","Sample e",
"Sample f","Sample g","Sample h","Sample i","Sample J"),
rotate.title=0)
dev.off()
}
Also, I am unable to plot the Ideogram track for Plasmodium Falciparum above the Genome axis track using the following code
>gen <- genome(GRanges(df))
>gen[is.na(gen)] <- "Pf"
>IdeogramTrack(genome = gen, chromosome = df$chrom[1])
Error in if (!token %in% base::ls(env)) { : the condition has length > 1
>df
chrom start end
1 PvP01_01_v2 1 1021664
2 PvP01_02_v2 1 956327
3 PvP01_03_v2 1 896704
4 PvP01_04_v2 1 1012024
5 PvP01_05_v2 1 1524814
6 PvP01_06_v2 1 1042791
7 PvP01_07_v2 1 1652210
8 PvP01_08_v2 1 1761288
9 PvP01_09_v2 1 2237066
10 PvP01_10_v2 1 1548844
11 PvP01_11_v2 1 2131221
12 PvP01_12_v2 1 3182763
13 PvP01_13_v2 1 2093556
14 PvP01_14_v2 1 3153402
15 PvP01_API_v2 1 29582
16 PvP01_MIT_v2 1 5989
## Also tried
> gen <- genome(GRanges(df[1,]))
> IdeogramTrack(genome = gen, chromosome = df$chrom[1])
Error in eval(expression, envir = callEnv) :
'NA' is not a valid UCSC genome.
> gen[is.na(gen)] <- "Pf"
> IdeogramTrack(genome = gen, chromosome = df$chrom[1])
Error in eval(expression, envir = callEnv) :
'Pf' is not a valid UCSC genome.
Feature Request: You can use above function or write a better function to make multi-sample-multi-chromosome-coverage plots. Besides, it will be really helpful to have a vignette on dealing with the non-model organism while using Gviz.
> sessionInfo( )
R version 4.2.1 (2022-06-23)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8
[4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] dplyr_1.0.9 Gviz_1.40.1 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[5] IRanges_2.30.0 S4Vectors_0.34.0 BiocGenerics_0.42.0
loaded via a namespace (and not attached):
[1] ProtGenerics_1.28.0 bitops_1.0-7 matrixStats_0.62.0
[4] bit64_4.0.5 filelock_1.0.2 RColorBrewer_1.1-3
[7] progress_1.2.2 httr_1.4.3 backports_1.4.1
[10] tools_4.2.1 utf8_1.2.2 R6_2.5.1
[13] rpart_4.1.16 lazyeval_0.2.2 Hmisc_4.7-0
[16] DBI_1.1.3 colorspace_2.0-3 nnet_7.3-17
[19] gridExtra_2.3 tidyselect_1.1.2 prettyunits_1.1.1
[22] bit_4.0.4 curl_4.3.2 compiler_4.2.1
[25] cli_3.3.0 Biobase_2.56.0 htmlTable_2.4.0
[28] xml2_1.3.3 DelayedArray_0.22.0 rtracklayer_1.56.1
[31] checkmate_2.1.0 scales_1.2.0 rappdirs_0.3.3
[34] stringr_1.4.0 digest_0.6.29 Rsamtools_2.12.0
[37] foreign_0.8-82 XVector_0.36.0 dichromat_2.0-0.1
[40] htmltools_0.5.2 base64enc_0.1-3 jpeg_0.1-9
[43] pkgconfig_2.0.3 MatrixGenerics_1.8.1 ensembldb_2.20.2
[46] dbplyr_2.2.1 fastmap_1.1.0 BSgenome_1.64.0
[49] htmlwidgets_1.5.4 rlang_1.0.3 rstudioapi_0.13
[52] RSQLite_2.2.14 BiocIO_1.6.0 generics_0.1.2
[55] BiocParallel_1.30.3 VariantAnnotation_1.42.1 RCurl_1.98-1.7
[58] magrittr_2.0.3 GenomeInfoDbData_1.2.8 Formula_1.2-4
[61] interp_1.1-2 Matrix_1.4-1 Rcpp_1.0.8.3
[64] munsell_0.5.0 fansi_1.0.3 lifecycle_1.0.1
[67] stringi_1.7.6 yaml_2.3.5 SummarizedExperiment_1.26.1
[70] zlibbioc_1.42.0 BiocFileCache_2.4.0 blob_1.2.3
[73] parallel_4.2.1 crayon_1.5.1 deldir_1.0-6
[76] lattice_0.20-45 Biostrings_2.64.0 splines_4.2.1
[79] GenomicFeatures_1.48.3 hms_1.1.1 KEGGREST_1.36.2
[82] knitr_1.39 pillar_1.7.0 rjson_0.2.21
[85] codetools_0.2-18 biomaRt_2.52.0 XML_3.99-0.10
[88] glue_1.6.2 biovizBase_1.44.0 latticeExtra_0.6-30
[91] data.table_1.14.2 png_0.1-7 vctrs_0.4.1
[94] gtable_0.3.0 purrr_0.3.4 assertthat_0.2.1
[97] cachem_1.0.6 ggplot2_3.3.6 xfun_0.31
[100] AnnotationFilter_1.20.0 restfulr_0.0.15 survival_3.2-13
[103] tibble_3.1.7 GenomicAlignments_1.32.0 AnnotationDbi_1.58.0
[106] memoise_2.0.1 cluster_2.1.3 ellipsis_0.3.2