
Hello!
I'm trying to do DGE on a small subset of data before importing and analyzing the rest. I'm new to R, and very new to DESeq2 (first time). I've been following the vignettes and tutorials as best I can.
I've been able to do "analysis" on my subset (2 controls and 2 treatments) without errors finally. However, upon analyzing the results there are 0 genes listed of interest.
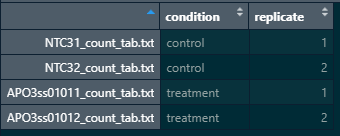
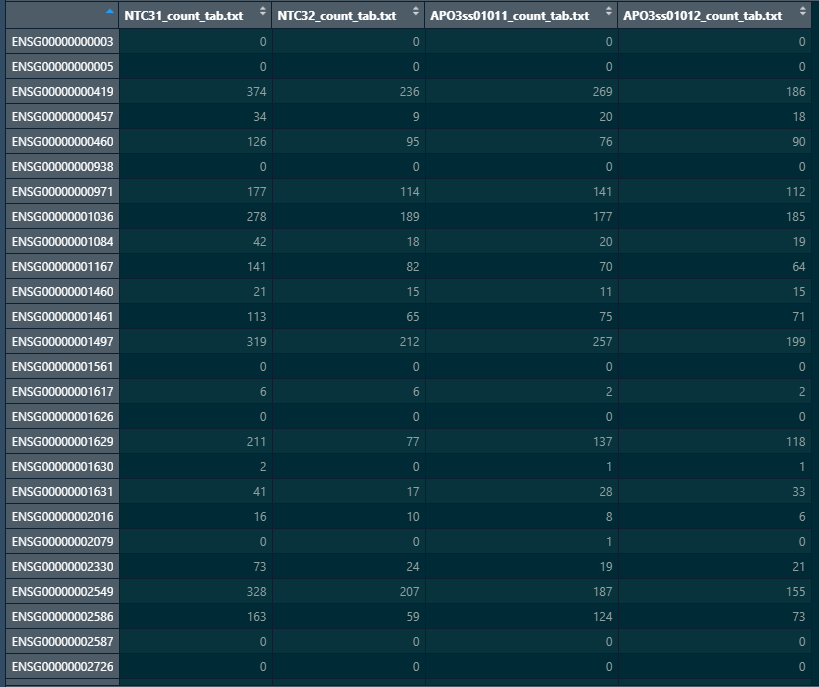
Here is a snapshot of my colData and count matrix
#matching coldata names with count matrix names
all(rownames(coldata_sample_simple) %in% colnames(simple_count))
[1] TRUE
#adding reference factor to base comparison on
dds$condition <- relevel(dds$condition, ref = "control")
#creating DESeq dataset object
dds_simple <- DESeqDataSetFromMatrix(countData = simple_count,
colData = coldata_sample_simple,
design = ~ condition)
dds_simple
class: DESeqDataSet
dim: 58740 4
metadata(1): version
assays(1): counts
rownames(58740): ENSG00000000003 ENSG00000000005 ... __not_aligned
__alignment_not_unique
rowData names(0):
colnames(4): NTC31_count_tab.txt NTC32_count_tab.txt APO3ss01011_count_tab.txt
APO3ss01012_count_tab.txt
colData names(2): condition replicate
summary(res_simple)
out of 19900 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 0, 0%
LFC < 0 (down) : 0, 0%
outliers [1] : 0, 0%
low counts [2] : 0, 0%
(mean count < 0)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
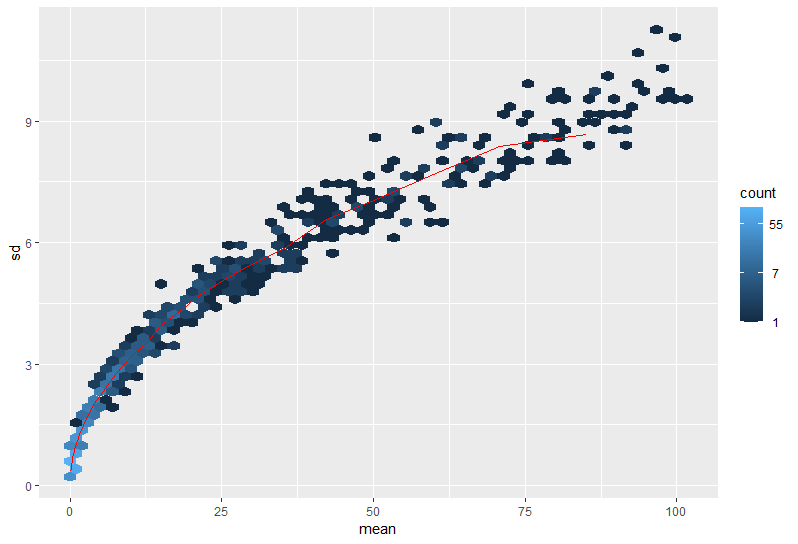
Here are a couple images of my data (from suggested analyses)
sessionInfo( )
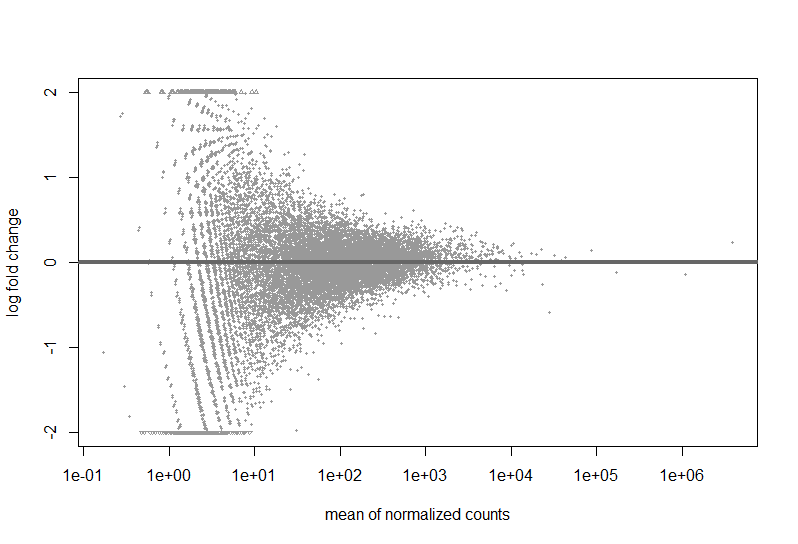
Just looking at the MAplot, I feel like there should be something that is differentially expressed. Do I need more samples to be able to make conclusions?
There are a tremendous amout of 'alignment not unqiue' reads. Would this contribute?
res_simple
log2 fold change (MLE): condition treatment vs control
Wald test p-value: condition treatment vs control
DataFrame with 58740 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000000003 0.0000 NA NA NA NA NA
ENSG00000000005 0.0000 NA NA NA NA NA
ENSG00000000419 258.0545 -0.110980 0.275095 -0.403425 0.686636 0.999944
ENSG00000000457 18.8728 0.264714 0.777033 0.340673 0.733350 0.999944
ENSG00000000460 95.2979 -0.122565 0.378462 -0.323851 0.746051 0.999944
... ... ... ... ... ... ...
__no_feature 1101333 -0.136970 0.163765 -0.836385 0.402938 0.999944
__ambiguous 169735 -0.115875 0.167525 -0.691687 0.489134 0.999944
__too_low_aQual 0 NA NA NA NA NA
__not_aligned 0 NA NA NA NA NA
__alignment_not_unique 3887524 0.232354 0.229656 1.011747 0.311659 0.999944
sum(res_simple$baseMean, na.rm=TRUE)
[1] 7969418
Thank you so much for taking the time and for any help!

I'd remove any of those "meta" features from the data.
Also, the
summaryshown in question looks like it was run before running the testing procedure, i.e.,Hi merv, Thanks so much for the advice and reply. I removed the metafeatures as suggested. I also re-ran the test and summary commands. Here is the output.
You simply have no DEGs at that sample size, your study is likely underpowered. The MA-plot suggests that based on the logFCs there are genes basically eligable to be DE but it's not significant at FDR<0.1. You need more samples. Check the PCA (
plotPCA()), see vignette, to see whether batch effects play a role.Thanks for the reply ATpoint, Okay, I will, thanks!