
I have samples originating from two tissues (brain and kidney). For a given tissue, I have cancer and normal samples. Brain = 13 (normal = 5 ; cancer = 8) Kidney = 17 (normal = 5 ; cancer = 12)
Goals
I want to identifying the DEGs comparing cancer between tissues and normal between tissues. For some patients, I have paired normal-cancer for a given tissue sample.
In addition to having confidence in the returned DEGs, I’d like to use the CPM-TMM normalized counts for other analyses outside voom.
Problem: The DEGs are biased for one direction. I don't think this is fully due to the biology.
I’m finding that if I take the housekeeping genes as done in the original TMM paper and compute the LFC, I’d expect to see something that’s 0 centered, but I’m seeing a shifter median LFC of -0.1 which for housekeeping genes is clearly wrong. Here, orange line is the observed median; blue, expected median; red line is gaussian with corresponding mean of orange line
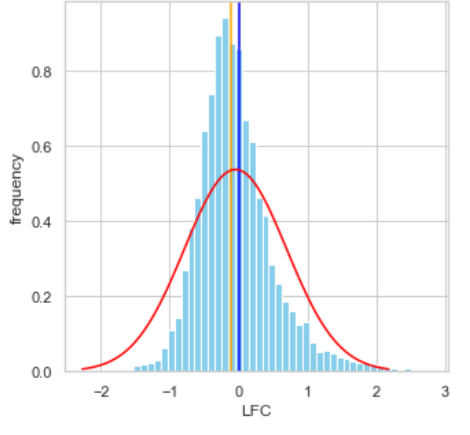
Gene Ontology of the returned DEGs yielded what I’d expect with the biology system and what I’ve confirmed with orthogonal analysis methods.
That said, I’m skeptical how attributable this is to the biology vs. technical artifact. And I can’t do analyses with the TMM-adjusted counts.
I'm very stuck for how to proceed.
What I’ve done:
I observed that I have some outliers on the MDS plot. I eliminated the outlier at (-2, 3) in subsequent analyses. Here’s the MDS plot:
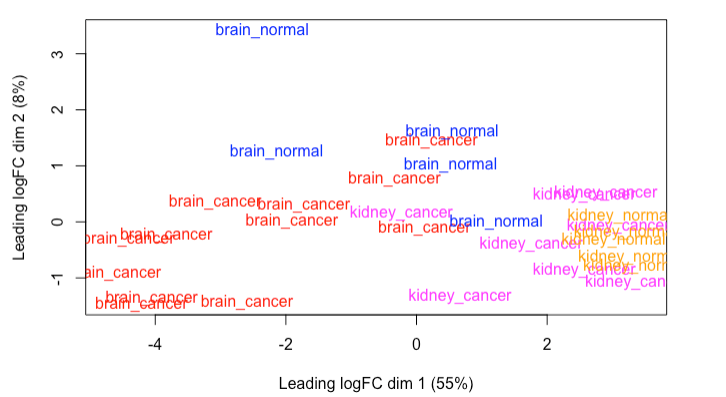
I recognize that the two cancer groups (notably brain_cancer) exhibits high in-group variation. There’s not much I can do about this other than I identified all plausible sources of variation by coloring the points accordingly assessing if I observed any pattern on any of the pairwise combinations of the glMDSPlot, I added it as a covariate to the design matrix.
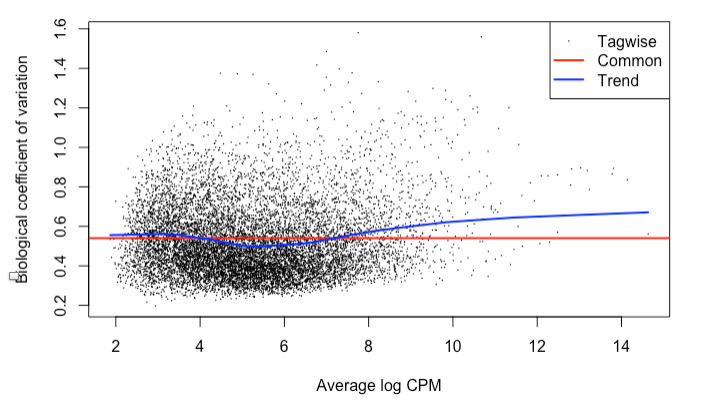
The BCV is 0.54 for the normal and cancer samples. I recognize this is high. The BCV for normal only is 0.34, which is generally consistent with what I’ve read for a BCV of ~0.4.
Since it's good practice from the RNAseq , I made MD plots for each sample. In my eyes, this is not zero-centered but rather shifted upwards ~ 2 on the y-axis. This was true of all but two of the samples. Here’s what I mean:
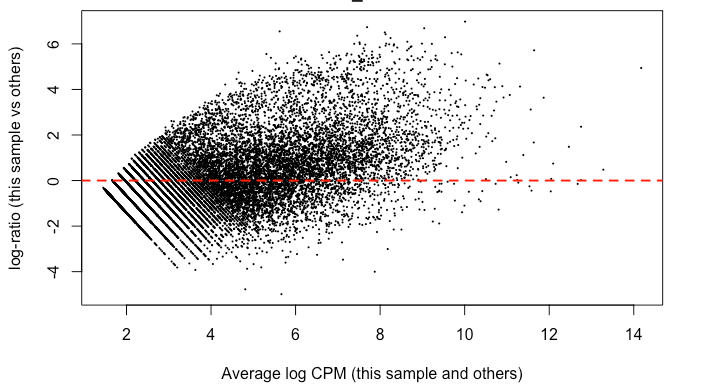
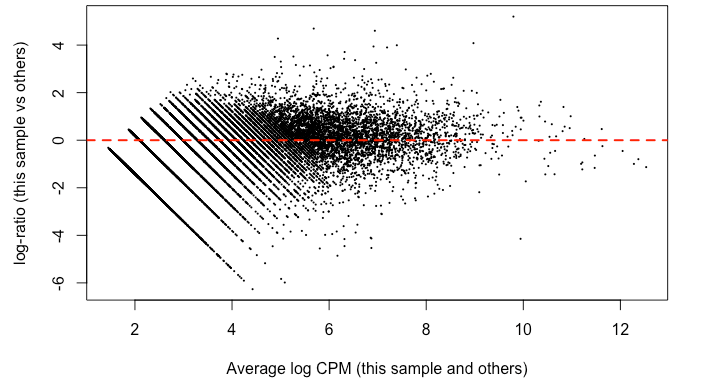
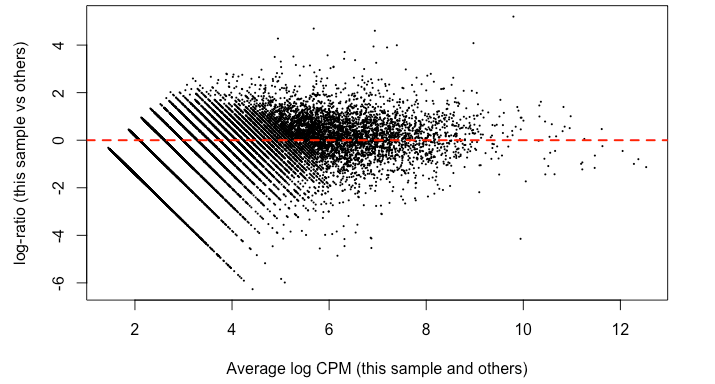
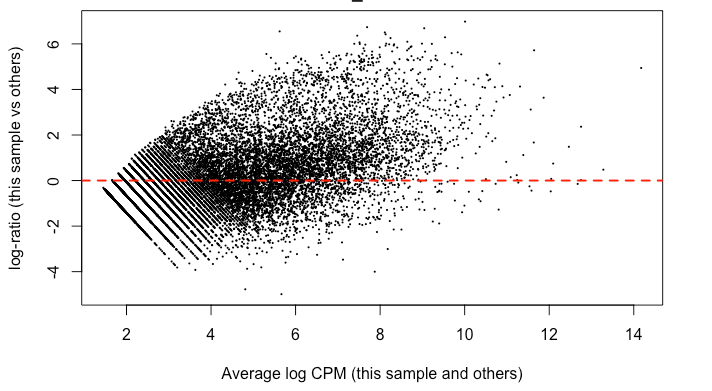
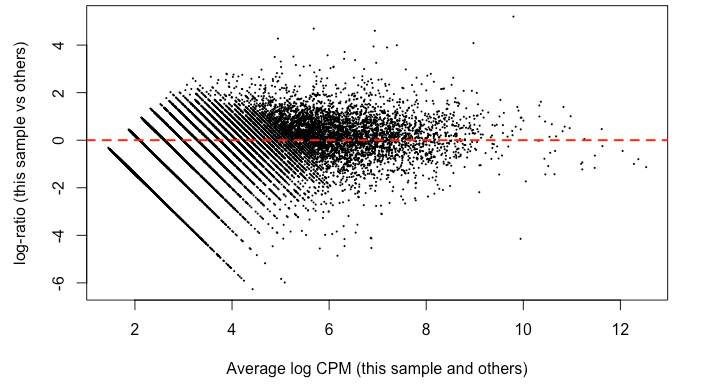
I find it highly alarming that there’s a small, but nonetheless persistent negative median LFC for housekeeping genes. It’s making me not trust the DEG. I tried filtering the CPM cutoff for pre-filtering genes going into the DEG but that didn't materially impact the mean-variance trend which comparing against the online guides doesn't seem too off
Questions: (1) Is there more I can do to address this persistent difference in the cancer samples? I was thinking of recentering all LFC based on the observed median from the HK genes for other comparisons I wanted to make... (2) Are the MD plots shifted or is it just me? (3) Is what I'm concerned about reasonable or is the median I'm worried about normal? I’m relatively new to voom, read a lot of documentation. I did not use voom() together with eBayes(trend=TRUE). I'm using voomWithQualityWeights given the ratio of the max to min library size. I tried filtering gene expression a priori as described in the Chen et al 2016 guide.
Thank you for creating such a well-document tool and supporting it. I learned from all the posts I read.
Code (if it's helpful):
counts <- read.csv(paste0("../", countsf), header = TRUE, row.names = 1)
meta <- read.csv(paste0("../", metaf),
header = TRUE)
dge <- dge[keep, , keep.lib.sizes = FALSE]
dge <- calcNormFactors(dge, method = 'TMM')
design <- model.matrix(~0 + meta$tissue + meta$disease_status+ meta$input_rna + meta$seq_batch + meta$sample_id, data = meta)
vwts <- voomWithQualityWeights(counts = dge,
design = design,
normalize.method = 'none',
method = "genebygene",
plot=TRUE)
corfit <- duplicateCorrelation(vwts, design, block = meta$sample_id)
vwts <- voomWithQualityWeights(counts = dge,
design = design,
normalize.method = 'none',
method = "genebygene",
block = meta$sample_id,
correlation = corfit$consensus,
plot=TRUE)
corAgain <- duplicateCorrelation(vwts, design, block = meta$sample_id)
vfit2 <- lmFit(vwts, design,
block = meta$sample_id,
correlation = corfit$consensus)
cm <- makeContrasts(
brain_cancer_normal = brain_cancer - brain_normal,
kidney_cancer_normal = kidney_cancer - kidney_normal,
levels = colnames(design)
)
vfit3 <- contrasts.fit(vfit2, cm)
vfit3 <- eBayes(vfit3, trend = FALSE, robust = TRUE)
vfit3_decideTests <- decideTests(vfit3, method = "global", p.value = 0.05)

Thank you, Gordon. I have two follow up questions:
voomWithQualityWeights(dge, normalize.method = ‘quantile’, ...)and eliminated thecalcNormFactorsline. However, I’m unable to recover the associated normalized counts. How can I recover the associated normalization factors withquantileandcyclic loessor the adjusted counts? Else, is extracting the associated cpm-log transformed normalized matrix in the following way equivalent to the counts matrix used byvoomWithQualityWeights? I understand thatnormalizeBetweenArraysis for single channel microarray data.Is this a plausible basis for sticking with TMM normalization?
As far as I can see, the analysis using TMM normalization was satisfactory. I don't see anything of particular concern in any of the figures you showed, considering the nature of the data. You found yourself that the DE results seemed appropriate and interpretable.
It isn't meaningful to export voom objects to non-limma analyses. This is because voom generates precision weights and non-limma software tools are very unlikely to be able to utilize precision weights properly. Instead I recommend that you use
cpmto export log-counts-per-million to other analysis tools. If you are using TMM normalization, thenIf you are using quantile normalization, then
For cyclic loess, just substitute
"cyclic.loess"for"quantile".I don't recommend normalizing by house-keeping genes, but you seem to have much more confidence in them than I do. You can normalize specifically to house-keeping genes either (1) by computing the TMM factors just for the house-keeping genes and then applying them to the whole analysis or (2) by using loess normalization and upweighting the housekeeping genes. Unfortunately the second method is not available as part of voom -- you would have to use limma-trend instead. Alternatively you could try RUVSeq to create surrogate variables from your housekeeping genes.
I don't want to comment on your lfc plots. I don't know which linear model you have fitted (your code gives two), or which contrast you are plotting, or whether the contrasts are the same in your first and second posts, or what the red density represents (it doesn't seem to match the histograms). Note that the TMM paper did not present a histogram of normalized logFC for house-keeping genes and did not claim that the normalized logFC would have median equal to zero. The TMM paper reported that a substantial proportion of the housekeeping genes were still DE after normalization.
You might be assuming that TMM normalization is intended to make the logFC average to zero, but the reality is different. The logFC will actually average to zero before TMM normalization. The purpose of TMM normalization is to move the logFC closer to zero for the main body of genes, even if that means moving the average logFC further from zero. For an extreme dataset like yours, the majority of genes are probably DE, which is a problem for the most normalization methods. For a dataset with extreme amount of DE, the normalization methods tend to error on the side of making the logFCs more symmetric that they ideally should be if you could normalize perfectly. That is especially true of quantile vs TMM or cyclic loess.