
Hi all,
I successfully ran DESeq2, though I am looking for differential abundance of microbial genera, not gene expression. However, duplicate genera (assigned to different species in my taxa table) are listed out and numbered instead of collapsing. I would like to collapse duplicate genera.
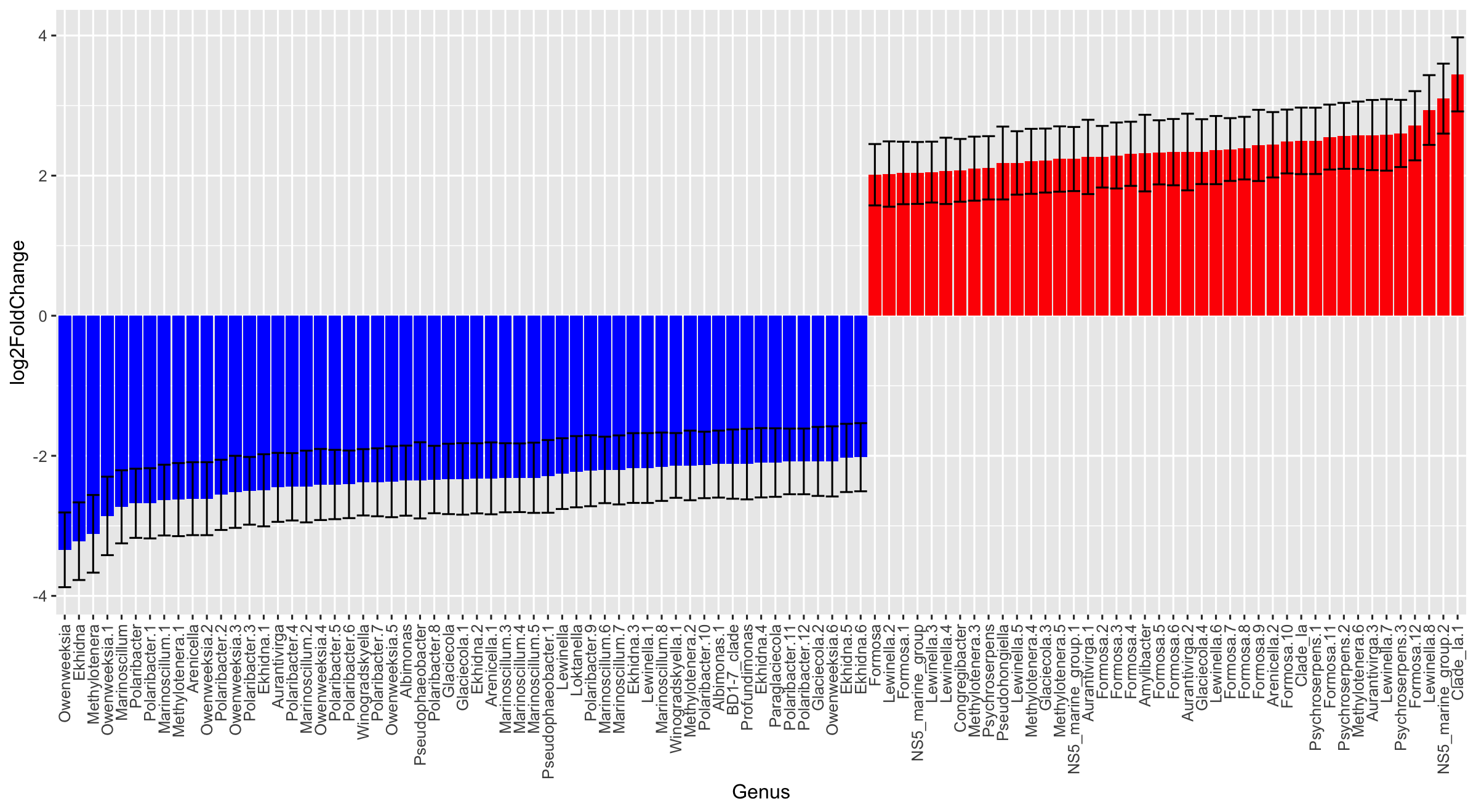
Here is what I ran to generate my figure:
```deg_plus1 <- transform_sample_counts(ds2, function(x) x+1) deg_deseq <- phyloseq_to_deseq2(deg_plus1, ~Sample.Type) DESEQ_deg <- DESeq(deg_deseq) res <- results(DESEQ_deg, tidy=TRUE)
To get table of results
sigASVs <- res %>% filter(padj<0.001 & abs(log2FoldChange)>2) %>% dplyr::rename(ASV=row) View(sigASVs)
Get only asv names
sigASVs_vec <- sigASVs %>% pull(ASV)
Prune phyloseq file
deg_DESeq <- prune_taxa(sigASVs_vec,deg_simple) sigASVs <- tax_table(deg_DESeq) %>% as.data.frame() %>% rownames_to_column(var="ASV") %>% right_join(sigASVs) %>% arrange(log2FoldChange) %>% mutate(Genus = make.unique(Genus)) %>% mutate(Genus = factor(Genus, levels=unique(Genus))) ggplot(sigASVs) + geom_bar(aes(x=Genus, y=log2FoldChange), stat="identity",fill=ifelse(sigASVs$log2FoldChange>0,"red","blue"))+ geom_errorbar(aes(x=Genus, ymin=log2FoldChange-lfcSE, ymax=log2FoldChange+lfcSE)) + theme(axis.text.x = element_text(angle=90, hjust=1, vjust=0.5)) ```
Many thanks!

How is your data before calculating the deg_DESeq? I see you use
phyloseq_to_deseq2which should work well, but have you double checked that each taxa is correctly summarized? Also note that DESeq2 is meant for low number of samples and raw counts and does many assumptions on the model used. It might be better to use a more general approach to calculate DEG.