Entering edit mode

Hello,
I had two questions regarding EPIC methylation array data analysis. I am using the minfi package for analysis, have removed low quality samples, processed the data with quantile normalization, and extracted both beta and m values.
- When examining MDS (or PCA) plots to examine sources of variation in the data, I see a large batch effect along PC2 corresponding to the plate in which the sample was sequenced, which I expected. However, there is a large unexplained difference between samples along the first principal component which is present in both plates. It does not correlate with position on the chip, and it does not have an association with age, sex, race, or biological condition. It accounts for a huge amount of variance (79%), so I was wondering if there are any common technical issues that people adjust for on the front end or if anyone has seen this discrepancy before.
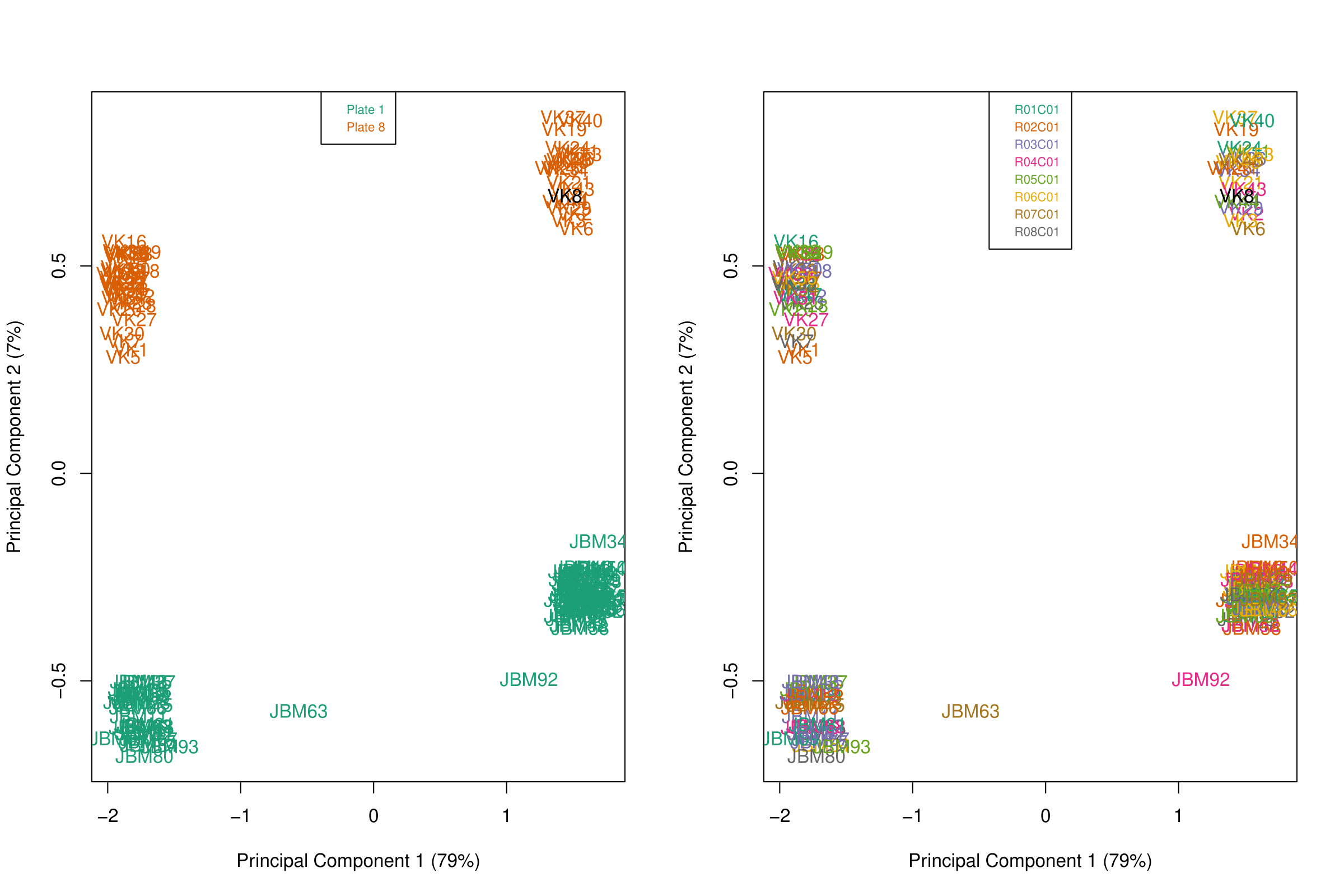
- Is linear regression (as in limma package) an appropriate model to use to determine differentially methylated CpGs/regions? The beta and M values have bimodal distributions which violates the normality assumption, I wasn't sure if another transformation was needed in order to achieve a normal distribution.
Thank you in advance for the help.
