Entering edit mode

Hi!
I use cluster function from CATALYST package as follows:
sce <- cluster(sce, features = "type",
xdim = 10, ydim = 10, maxK = maxk, seed = 1)
Each time I run the above function in a new R session I get slightly different results (e.g. percentages, clusters) using plotExprHeatmap
plotExprHeatmap(sce, features = "type", row_clust = TRUE, col_clust = FALSE,
by = "cluster_id", k = kmeta,
bars = TRUE, perc = TRUE)
.
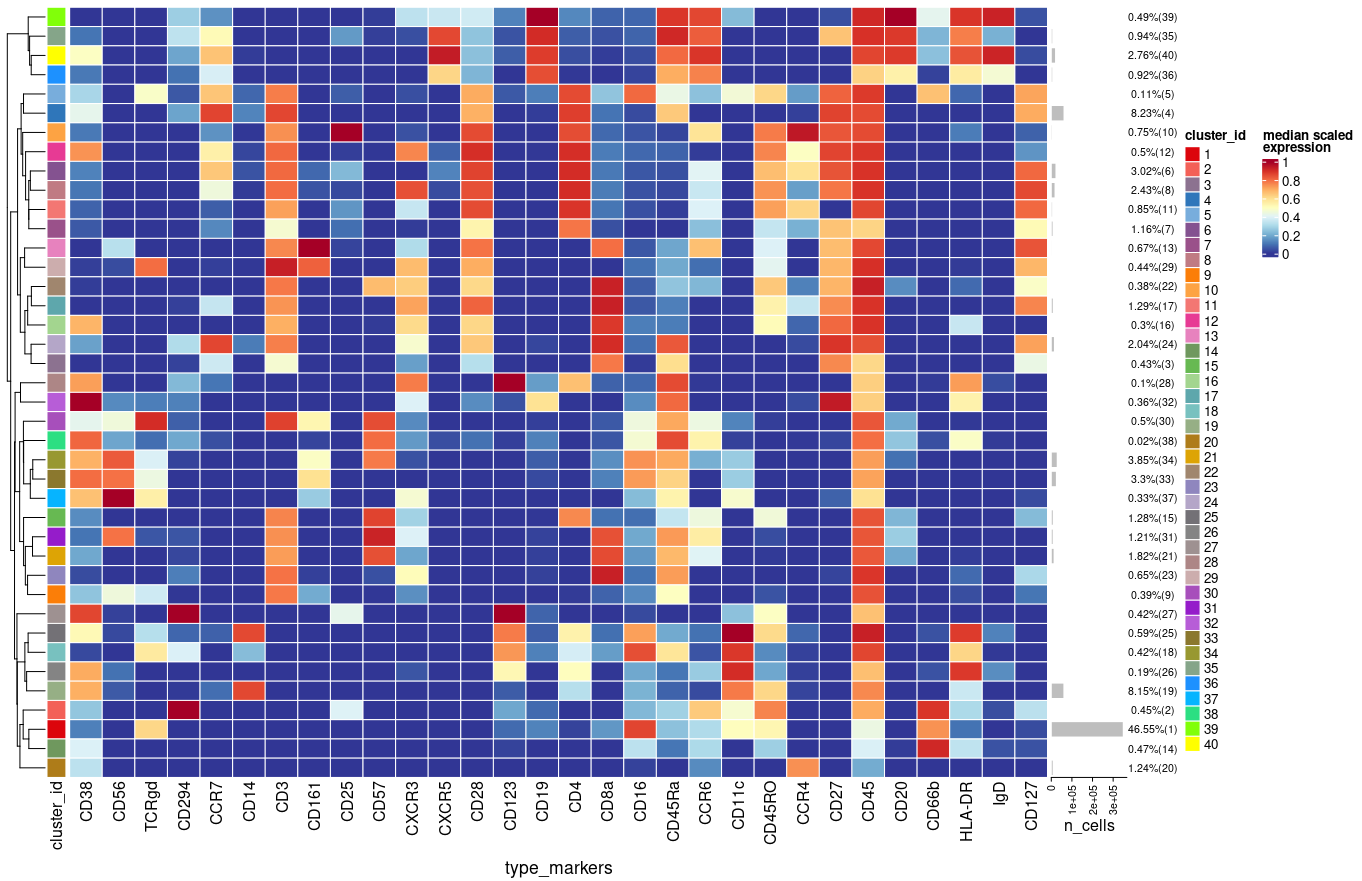
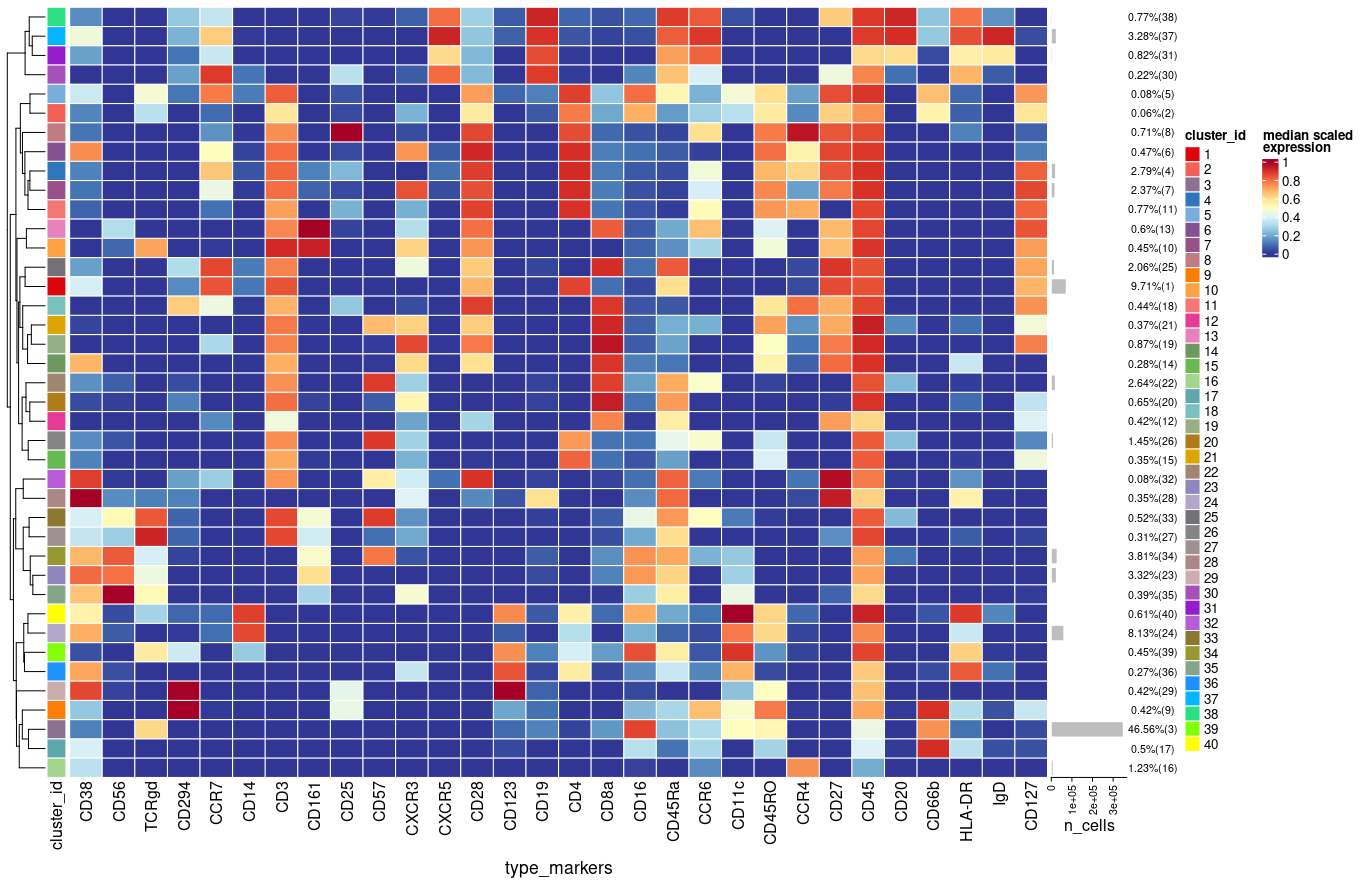
Is there any reason/explanation for that?
Thanks!
sessionInfo( )
R version 4.3.2 (2023-10-31)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: Europe/Athens
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] cowplot_1.1.1 ggplot2_3.4.4 writexl_1.4.2 CATALYST_1.24.0
[5] SingleCellExperiment_1.22.0 SummarizedExperiment_1.30.2 Biobase_2.60.0 GenomicRanges_1.52.0
[9] GenomeInfoDb_1.36.1 IRanges_2.34.1 S4Vectors_0.38.1 BiocGenerics_0.46.0
[13] MatrixGenerics_1.12.2 matrixStats_1.1.0 flowCore_2.12.2 readxl_1.4.3
loaded via a namespace (and not attached):
[1] bitops_1.0-7 gridExtra_2.3 sandwich_3.0-2 rlang_1.1.2
[5] magrittr_2.0.3 clue_0.3-65 GetoptLong_1.0.5 multcomp_1.4-25
[9] scater_1.28.0 ggridges_0.5.4 compiler_4.3.2 DelayedMatrixStats_1.22.6
[13] png_0.1-8 vctrs_0.6.4 reshape2_1.4.4 stringr_1.5.0
[17] pkgconfig_2.0.3 shape_1.4.6 crayon_1.5.2 backports_1.4.1
[21] XVector_0.40.0 scuttle_1.10.3 utf8_1.2.4 ggbeeswarm_0.7.2
[25] purrr_1.0.2 beachmat_2.16.0 zlibbioc_1.46.0 DelayedArray_0.26.7
[29] BiocParallel_1.34.2 tweenr_2.0.2 irlba_2.3.5.1 broom_1.0.5
[33] parallel_4.3.2 cluster_2.1.5 R6_2.5.1 stringi_1.7.12
[37] RColorBrewer_1.1-3 car_3.1-2 cellranger_1.1.0 Rcpp_1.0.11
[41] iterators_1.0.14 zoo_1.8-12 nnls_1.5 splines_4.3.2
[45] Matrix_1.6-1 igraph_1.5.1 tidyselect_1.2.0 viridis_0.6.3
[49] rstudioapi_0.15.0 abind_1.4-5 doParallel_1.0.17 codetools_0.2-19
[53] lattice_0.22-5 tibble_3.2.1 plyr_1.8.9 withr_2.5.2
[57] Rtsne_0.16 survival_3.5-7 polyclip_1.10-6 ConsensusClusterPlus_1.64.0
[61] circlize_0.4.15 pillar_1.9.0 ggpubr_0.6.0 carData_3.0-5
[65] foreach_1.5.2 generics_0.1.3 colorRamps_2.3.1 RCurl_1.98-1.12
[69] sparseMatrixStats_1.12.2 munsell_0.5.0 scales_1.2.1 gtools_3.9.4
[73] glue_1.6.2 tools_4.3.2 BiocNeighbors_1.18.0 ggnewscale_0.4.9
[77] data.table_1.14.8 ScaledMatrix_1.8.1 ggsignif_0.6.4 mvtnorm_1.2-2
[81] XML_3.99-0.15 Cairo_1.6-2 grid_4.3.2 plotrix_3.8-4
[85] tidyr_1.3.0 RProtoBufLib_2.12.1 colorspace_2.1-0 GenomeInfoDbData_1.2.10
[89] beeswarm_0.4.0 BiocSingular_1.16.0 ggforce_0.4.1 vipor_0.4.5
[93] rsvd_1.0.5 cli_3.6.1 fansi_1.0.5 viridisLite_0.4.2
[97] cytolib_2.12.1 S4Arrays_1.0.4 ComplexHeatmap_2.16.0 dplyr_1.1.3
[101] gtable_0.3.4 rstatix_0.7.2 digest_0.6.33 FlowSOM_2.8.0
[105] ggrepel_0.9.4 TH.data_1.1-2 rjson_0.2.21 drc_3.0-1
[109] farver_2.1.1 lifecycle_1.0.4 GlobalOptions_0.1.2 MASS_7.3-60