Entering edit mode

I want to know which methods are commonly used for RNA seq data if the goal is to identify monotonic increasing or decreasing gene expression over time or across stages using the results of differential gene expression analysis?

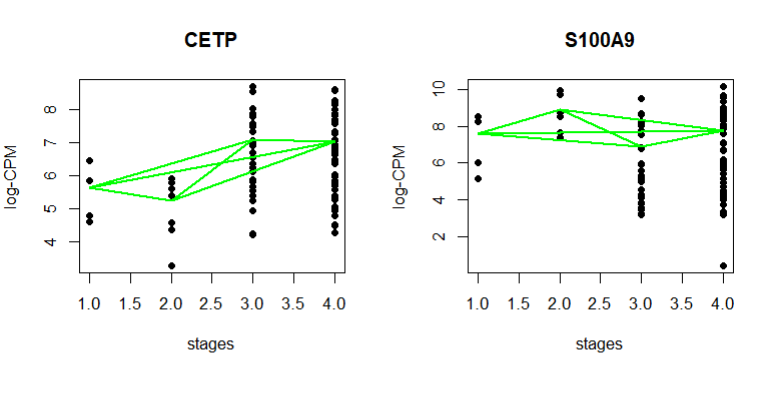
Cross-posted to Biostars https://www.biostars.org/p/9585490/