
Hello,
We are trying to find the root issue of why we are having differing final output results from using the same datasets, parameters, functions, R (4.4.1), RStudio ("2024.04.2+764"), and goseq (1.56.0) package versions. Using the code below, I have tried to replicate the results with the exact same cloned repository on multiple computers and operating systems while checking that all steps are identical when running. However, with this code I am receiving 31 significantly enriched terms, while when someone else clones the repository or the code is used on different operating systems we see there are no significantly enriched terms. This is clearly a major issue and we have tried multiple steps to solve the issue as updating all versions to be the same, trying on multiple systems, cloning and recloning the repo, updating the repo and confirming it has been pushed to Github correctly but we have run out of troubleshooting options. Any ideas or help would be much appreciated!
### Generate vector with names of all genes detected in our dataset
ALL.vector <- c(filtered_Pverr.annot$gene_id)
### Generate length vector for all genes
LENGTH.vector <- as.integer(filtered_Pverr.annot$length)
### Generate vector with names in just the contrast we are analyzing
ID.vector.down <- DEG.res %>%
filter(direction=="Downregulated") %>%
pull(gene_id)
##Get a list of GO Terms for all genes detected in our dataset
GO.terms <- filtered_Pverr.annot%>%
dplyr::select(gene_id, GO.ID)
##Format to have one goterm per row with gene ID repeated
split <- strsplit(as.character(GO.terms$GO.ID), ";")
split2 <- data.frame(v1 = rep.int(GO.terms$gene_id, sapply(split, length)), v2 = unlist(split))
colnames(split2) <- c("gene_id", "GO.ID")
GO.terms<-split2
##Construct list of genes with 1 for genes in contrast and 0 for genes not in the contrast
gene.vector.down=as.integer(ALL.vector %in% ID.vector.down)
names(gene.vector.down)<-ALL.vector#set names
#weight gene vector by bias for length of gene: load in the gene.vector(a list of all genes with 1 indicating it is a DEG in the group of interest and 0 meaning it is not a DEG) and bias.data (list of lengths for all genes)
pwf.down<-nullp(gene.vector.down, bias.data=LENGTH.vector)
data(genes)
pwf <- nullp(genes, 'hg19', 'ensGene')
#run goseq using Wallenius method for all categories of GO terms: load in the pwf object (gene name, a 1 or 0 to indicate DEG, the length, and the pwf function; generated from pwf above), and the list of GO terms for all genes
GO.wall.down<-goseq(pwf.down, gene2cat=GO.terms, test.cats=c("GO:BP", "GO:MF", "GO:CC"), method="Wallenius", use_genes_without_cat=TRUE)
GO.down <- GO.wall.down[order(GO.wall.down$over_represented_pvalue),]
colnames(GO.down)[1] <- "GOterm"
#adjust p-values
GO.down$bh_adjust <- p.adjust(GO.down$over_represented_pvalue, method="BH") #add adjusted p-values
#Filtering for p < 0.05
GO.down <- GO.down %>%
dplyr::filter(bh_adjust<0.05) %>%
dplyr::arrange(., ontology, bh_adjust)
Output of data from MacOS Monterey version 12.6.6:
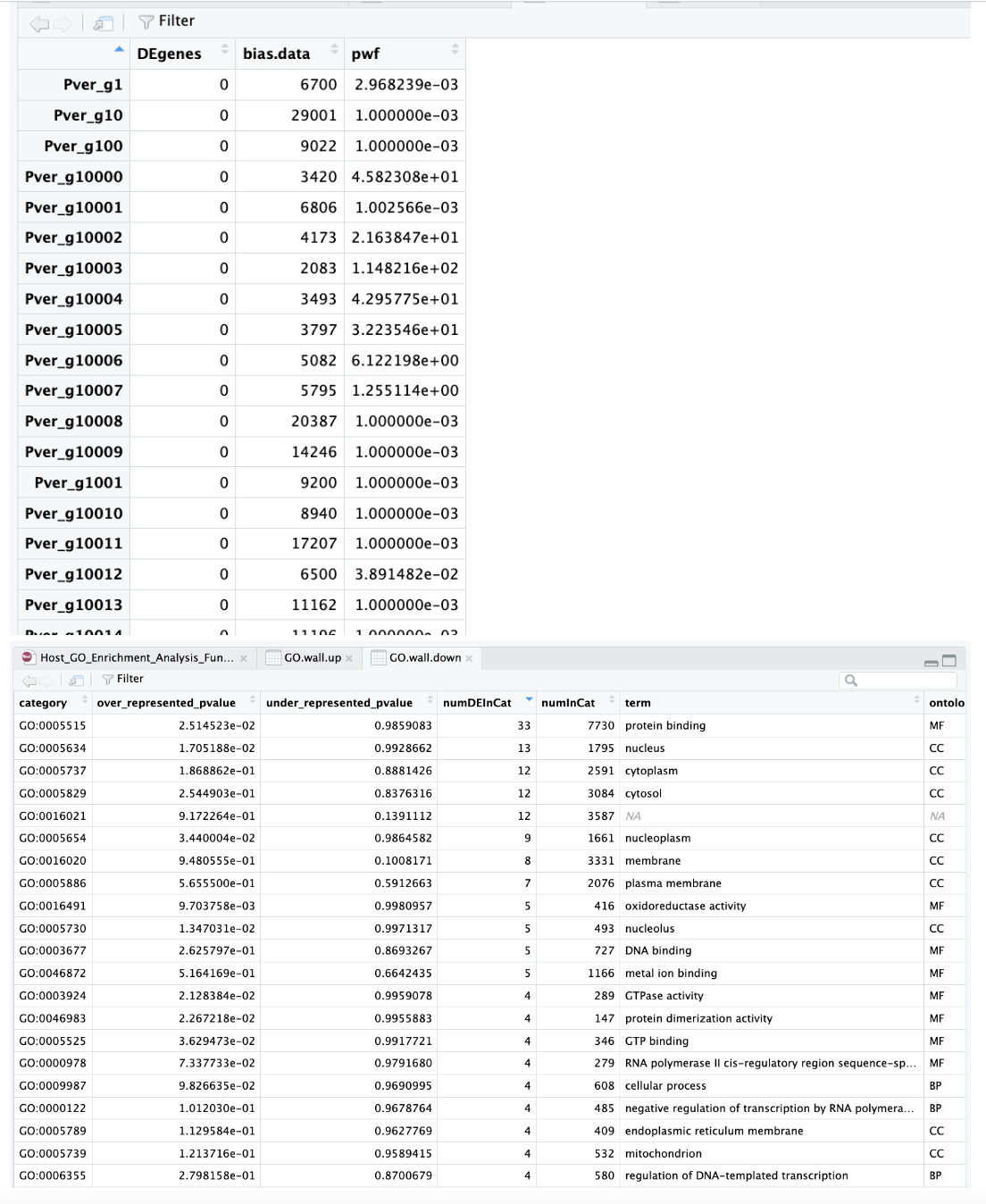
Output of data from macOS Sonoma 14.5:
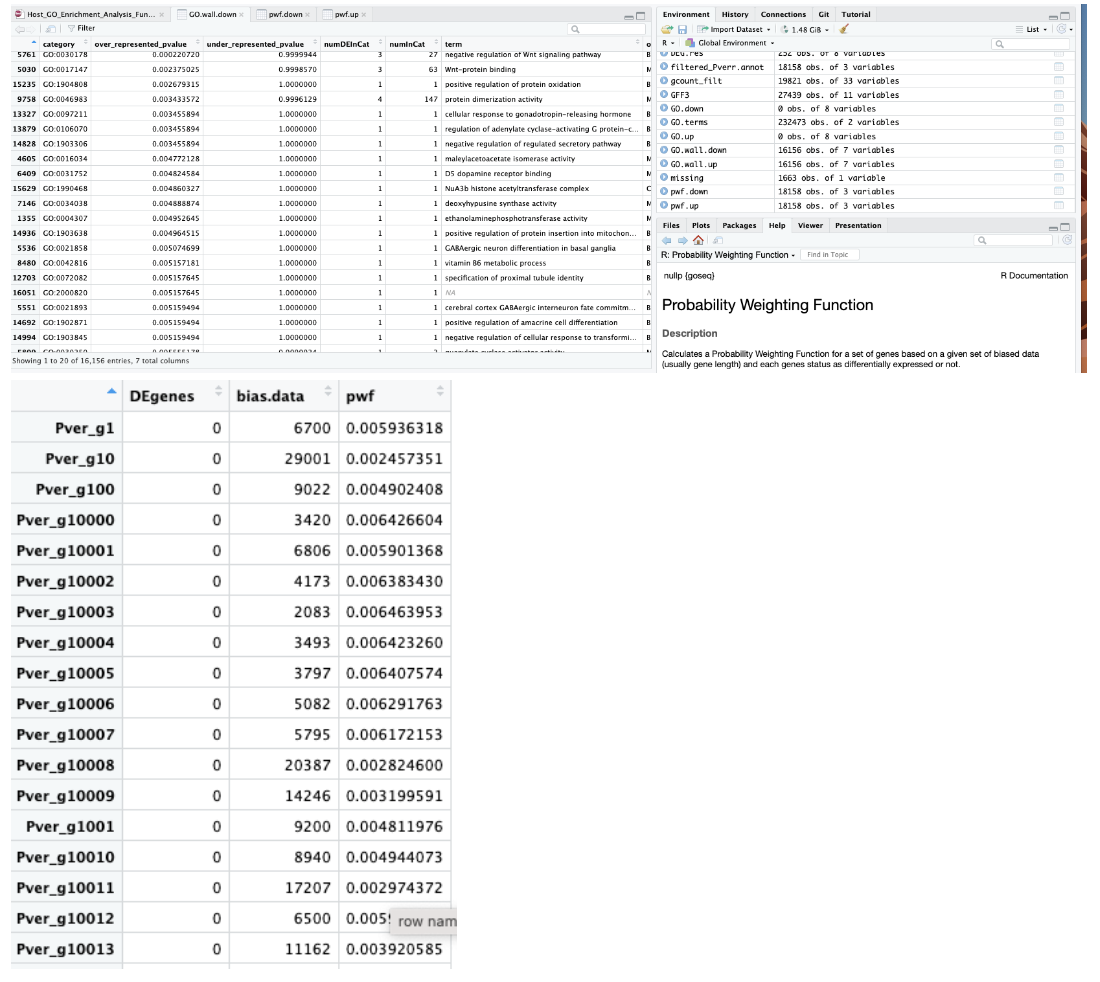
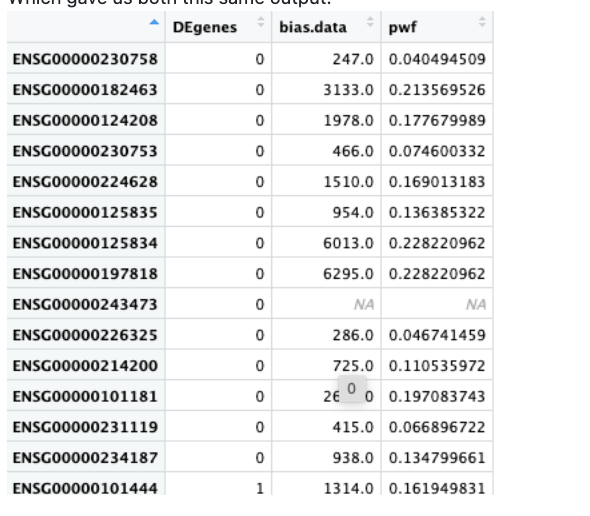
We also both ran a test dataset:
data(genes)
pwf <- nullp(genes, hg19, ensGene)
Which gave us both this same output:


Hi Federico,
Thank you for the quick response.
Yes, we made sure that all softwares and updates to packages and R and Rstudio were all the same. Also, between each of us we are using the exact same code and repositories for the data storage and download.
Please feel free to download the data and full repository from GitHub to see what results you get. Here is the GitHub link: https://github.com/hputnam/Becker_E5/blob/master/RAnalysis/Scripts/RNA-seq/Host/Host_GO_Enrichment_Analysis_Func_Annot.Rmd
Hi Danielle, I managed to clone the repo. Thanks for sharing that!
On my machine, MacOS 12.7, I have the following results:
nullp()... with a fairly odd shape in the fit plot for the output of
nullp()- not something that plateaus nicely "as it should".Maybe because you don't have so many DE genes?
So... on the line of your 12.6 setting.
goseq()Running then
goseq()I have this (many lines included to have thenegative regulation of Wnt signaling pathwayyou are showing in the Sonoma output)The output of
GO.downyou are showing for Monterey can probably not be in sync with your screenshots. I am expecting that if the overrepresented p value is not that small, upon correction this should just be filtered out - and here you are showing me some (fairly generic) hits (protein binding, cytosol, ...).All in all my output seems to resemble more the Sonoma-flavored analysis. And this, even if my
pwflooked more... monterey-like.--> I am kinda confused myself with the mix-up of results.
Can you try to run an alternative method on this very dataset, provided it fits with the custom gene-go annotation you need to provide? It can be that clusterProfiler can support that, not too sure if topGO can also cover this for you (I think so, but have no experience on such flavors).
Sorry I could not get to the solution, but maybe it can point you to an alternative at least? I agree that the differences are at least... odd to see.
HTH,
Federico
For completeness, this is my machine now:
... which is basically the latest release + the devel version of
goseqHi Federico,
Thank you for running that on your machine. You did get consistent results with my dataset when I ran it but that is because we both had run this on the same machine. I think the larger issue that still needs to be solved with goseq and its functions is that this is not consistent when run on a different machine. Which is very concerning when data analyses and the results we publish need to be reproducible for anyone that moves forward or has used goseq for enrichment analyses in the past. We are looking into a non-model organism (coral) which has not been available for clusterProfiler or topGO in the past but I can look further into these alternative options. Please let me know if you think anything can be amended in goseq to address these issues.
I might be too less aware of the internals - especially regarding the ones in cases where there seems to be not too much as DE to have a "good estimate" of the length bias relationship (too few points in the bins, I fear).
But you are right - the odd and worrisome thing is the change of behavior across OS versions (at least, this is what I got with mine). It could be interesting to replicate this on a recent MacOS version. I'll see around a bit if someone with a recent enough version of that could chime in.