
I am trying to get a more complete annotation for EPICv2. The annotation from Illumina "Infinium MethylationEPIC v2.0" (link below), has 614365 out of 937055 probes not having information on associated gene and a lot of "unknowns" when it comes to gene features such as TSS1500 or 5UTR, as seen on picture below. When I check the position of CpG on UCSC I can find related gene. For example, cg06979118 is overlapped with SHANK2 gene, but has no gene annotation in "Infinium MethylationEPIC v2.0".
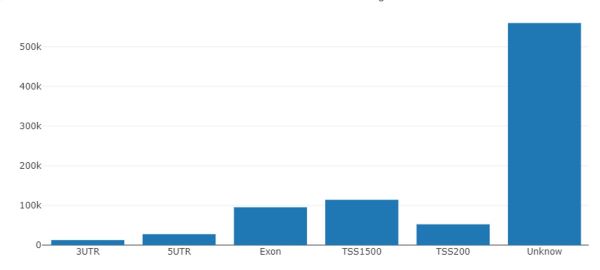
I followed ChAMP tutorial for analysis, that uses ChAMPdata (v2.31.1) package, which loads up the annotation based on "Infinium MethylationEPIC v2.0". The newer and developmental ChAMPdata versions do not have annotation for EPICv2 at all.
I have few questions
1) Has anyone used ChAMP and was able to load more complete annotation for EPICv2, meaning to find ChAMPdata package with complete annotation for EPICv2, since functions in ChAMP call data("probe.features.epicv2") even in champ.GSEA,
2) I have a half solution to my problem, I can annotate DMP after I find them but even then I am still not sure where to get a complete annotation. I can overlap probes used for EPICv1 with probes used for EPICv2. But then I am not sure how to perform GSEA analysis in this case since champ.GSEA calls data("probe.features.epicv2"), even though I provide it with DMPs and DMRs. Maybe someone could suggest the pipeline for that?
Infinium MethylationEPIC v2.0: https://support.illumina.com/array/array_kits/infinium-methylationepic-beadchip-kit/downloads.html
Tutorial for ChAMP: https://github.com/YuanTian1991/ChAMP-DemoRun/blob/main/EPICv2/illumina_demo_data_iScan/main.md

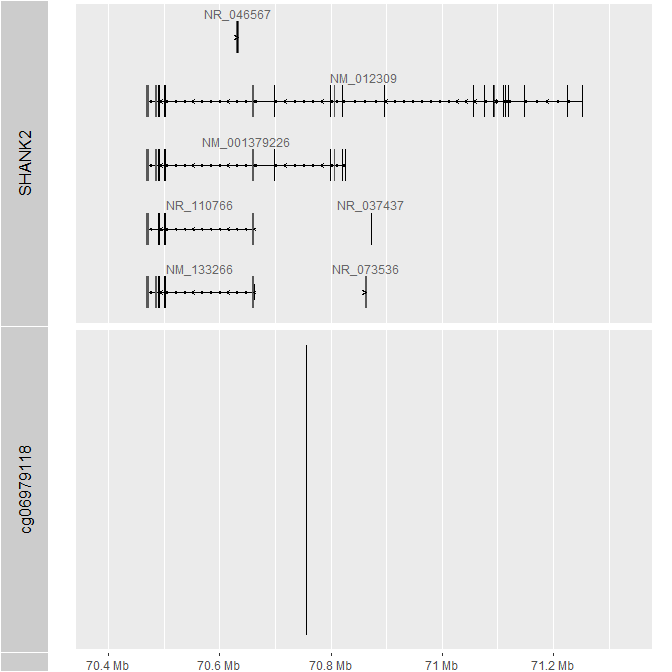

Don't know if any of these are of use or if these are what ChAMP uses under the hood:
IlluminaHumanMethylationEPICv2anno.20a1.hg38
IlluminaHumanMethylationEPICv2manifest
EPICv2manifest
Thank you for your reply.
I welcome any help I can get)
I checked the first two are based on the same file, Infinium MethylationEPIC v2.0
The last one seems to be also based on the same file, but it has more columns with more information