Entering edit mode

I have an annotation package, and I am not able to install the package locally. Since the package publishing takes more time, I want to use the package in locally. The annotation files were uploaded to cloud storage already. I am getting the below error,
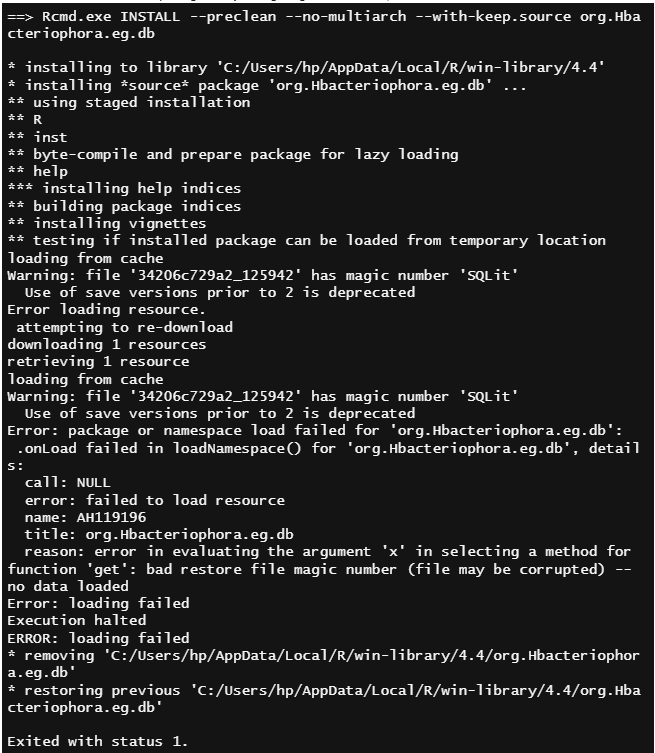
and this is the R script of the package
datacache <- new.env(hash=TRUE, parent=emptyenv())
org.Hbacteriophora.eg <- function() showQCData("org.Hbacteriophora.eg", datacache)
org.Hbacteriophora.eg_dbconn <- function() dbconn(datacache)
org.Hbacteriophora.eg_dbfile <- function() dbfile(datacache)
org.Hbacteriophora.eg_dbschema <- function(file="", show.indices=FALSE) dbschema(datacache, file=file, show.indices=show.indices)
org.Hbacteriophora.eg_dbInfo <- function() dbInfo(datacache)
org.Hbacteriophora.egORGANISM <- "Heterorhabditis bacteriophora"
.onLoad <- function(libname, pkgname) {
## Load AnnotationHub
hub <- AnnotationHub:::AnnotationHub()
## Query for the specific database (organism & sqlite)
query_result <- AnnotationHub:::query(hub, c("Heterorhabditis", "bacteriophora", "sqlite"))
## Assuming the SQLite file is the first query result
sqliteFile <- query_result[[1]]
## Assign the database file path and connection
dbfile <- sqliteFile$path
assign("dbfile", dbfile, envir=datacache)
dbconn <- dbFileConnect(dbfile)
assign("dbconn", dbconn, envir=datacache)
## Create the OrgDb object from the AnnotationHub resource
sPkgname <- sub(".db$", "", pkgname)
db <- loadDb(dbfile, packageName = pkgname)
dbNewname <- AnnotationDbi:::dbObjectName(pkgname, "OrgDb")
ns <- asNamespace(pkgname)
assign(dbNewname, db, envir=ns)
namespaceExport(ns, dbNewname)
packageStartupMessage(AnnotationDbi:::annoStartupMessages("org.Hbacteriophora.eg.db"))
}
.onUnload <- function(libpath) {
dbFileDisconnect(org.Hbacteriophora.eg_dbconn())
}
Please help me to solve this error.

Thank you for your suggestion. But I am getting the same error message while running this script.
OK, that appears to be a busted resource. Lori will have to look into it.
If that's the case the resource came from@KABILAN so they'll have to regenerate.
It means I have to upload all the files again. Isn't it?
Yes It appears to be an issue with the file.
No, you have to fix the package first. You are apparently trying to
loada SQLite DB as if it were a serialized R object. That's not howOrgDbpackages work. They are essentially a SQLite DB and some wrapper functions that perform SQL queries in order to extract data. Your code is trying to useloadto bring the SQLite DB into memory, which isn't a thing. In other words, consider this:That file (28a862ef49b5_125942) is an SQLite DB in my
hubCache, and is meant to be queried using SQL queries:And if I do the same with an existing
OrgDb, you can see it's very similar:I tried to read the vignette for
HubPub, but it is rather impenetrable IMO. The gist appears to be that you generate a regular package, put the SQLite DB file in the cloud or another publicly available place, and add a bit of extra stuff to the package to say where the DB is? Ideally you would be able to get anotherOrgDbfromAnnotationHuband just emulate what's in it, but I don't know how that is done?