
Working with Spatial Transcriptomics data (Large Spatial Experiment)
I have observed that while performing dimensionality reduction as the following:
speBreast <- runPCA(speBreast,
subset_row = sel)
and then:
speBreast <- runUMAP(speBreast,
dimred = 'PCA')
I obtain a certain result with the functions buildSNNGraph and igraph::cluster_leiden for the clustering of my samples.
k <- 10
g <- buildSNNGraph(speBreast,
k = k,
use.dimred = 'PCA')
k <- igraph::cluster_leiden(g,
objective_function = 'modularity',
resolution = 1.2)
However if I omit runUMAP() I obtain a completely different clustering result, although buildSNNGraph uses use.dimred = 'PCA' I obtain a substantially different clustering.
I believe it is caused by runUMAP() since when I change its dimred parameter from 'PCA' to NULL I obtain the exact same clustering of omitting runUMAP() entirely, as previously described.
Attached are the results of the Leiden clustering (right subplot) for runUMAP with dimred = 'PCA' and runUMAP with dimred = NULL respectively.
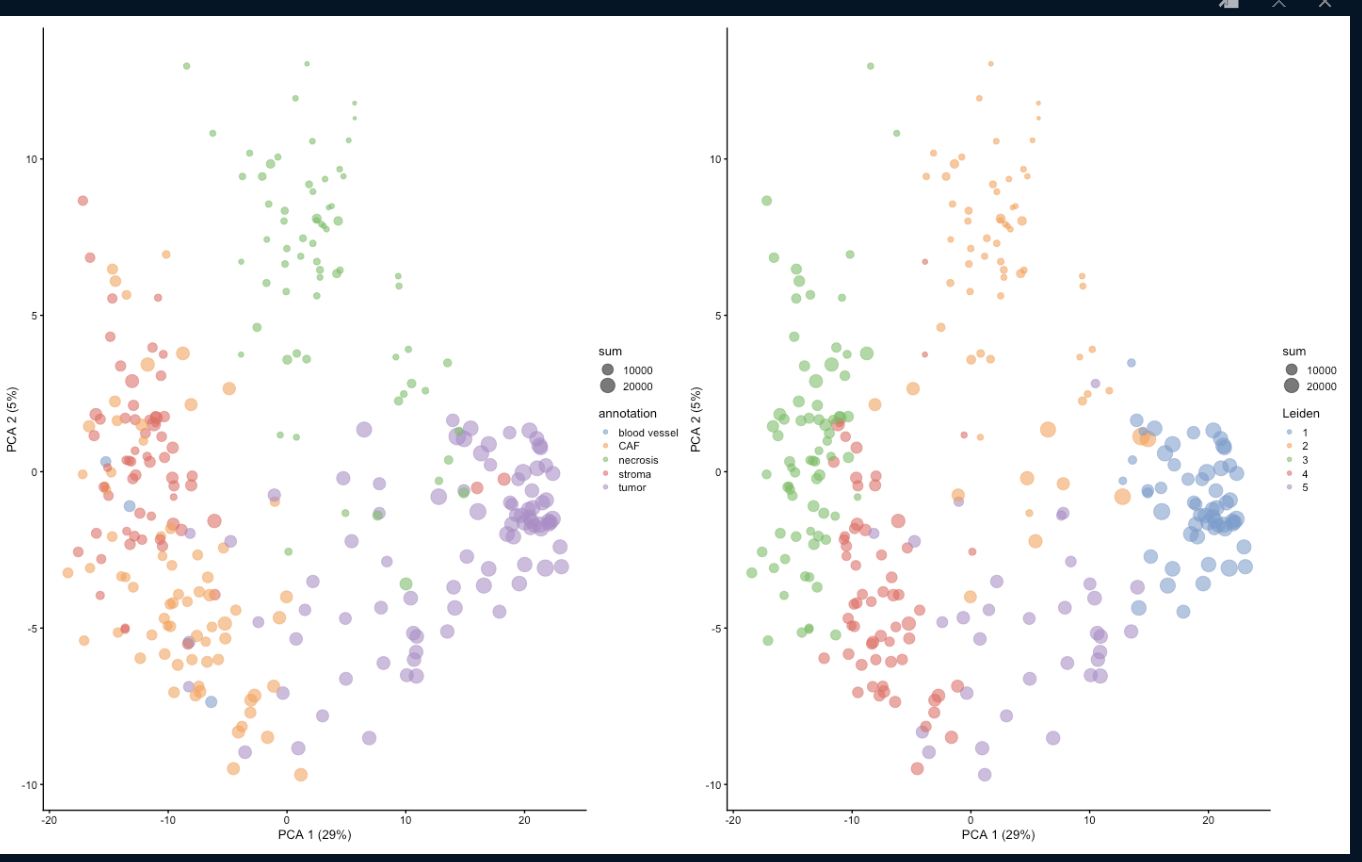
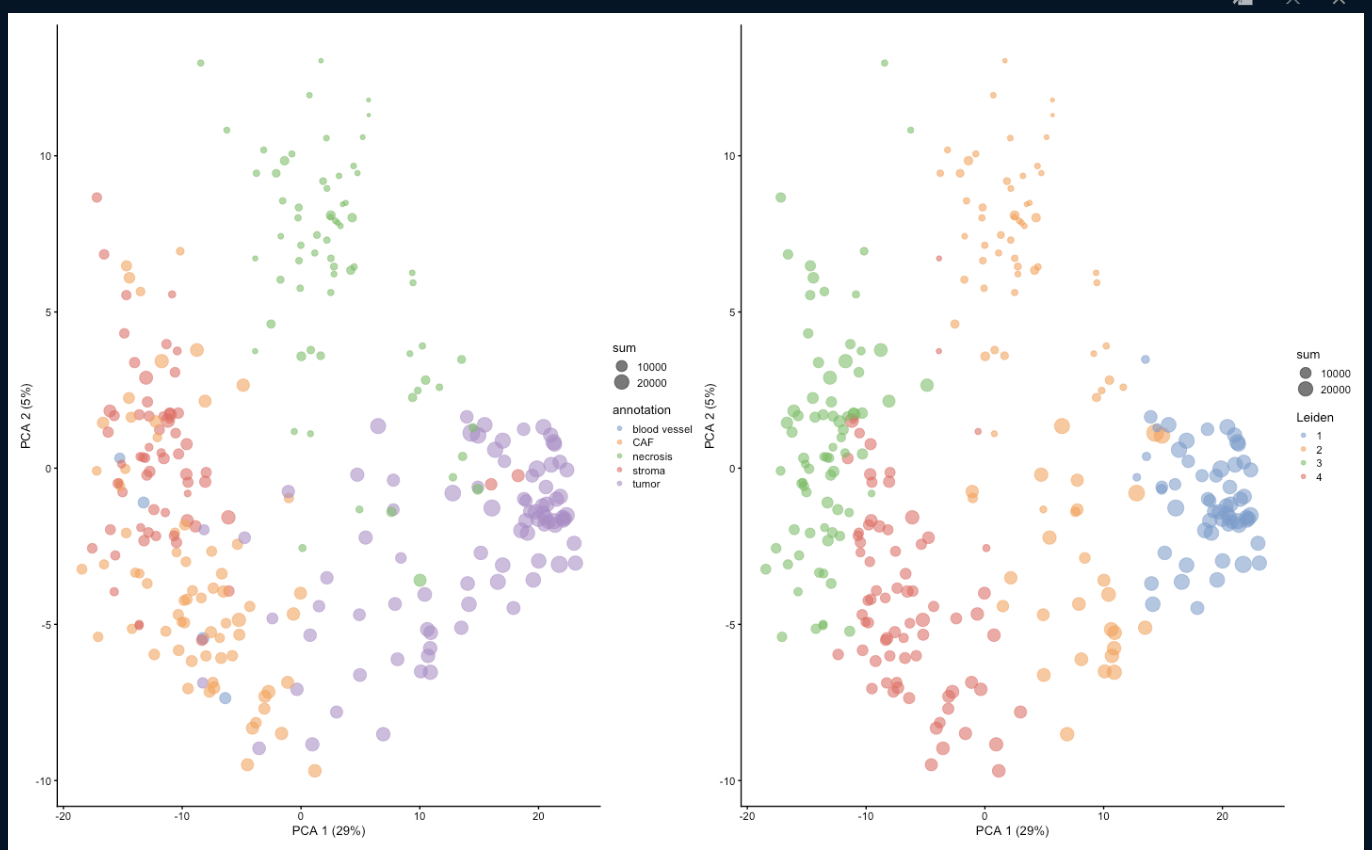
Any help is greatly appreciated

Just played with this a bit and I think it comes down to not having a fixed seed before running the leiden thingy. Same goes for PCA and UMAP, you should get used to always fix seeds before running such functions. Please try with fixed seeds and validate the issue is beyond that.