
Hi! I have two different bulk RNA-seq datasets that I want to compare (merge into one) and ideally perform differential expression analysis by using DESeq2. The problem is that altghough the samples were sequenced on the same platform, the sequencing depths of these datasets are very different (the first dataset has ca 2 million reads/sample; the second one ca 10x more).
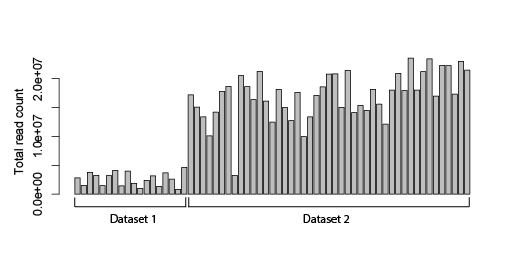
I am wondering if there is any method to adequately normalize the datasets, so they could be more comparable? Maybe based on some housekeeping genes or any other alternatives? I know that running data through DESeq2 already normalizes your data based on sequencing depth and RNA composition, but I still see a huge dataset-inflicted difference on a PCA plot (see plot below). And if I check the results file after DESeq2 normalization, some standard housekeeping genes (such as GAPDH or ACTB) show ca 10x difference between the samples in different datasets. See the example below (PC1 constitutes 90% difference, whereas PC2 only 4%).
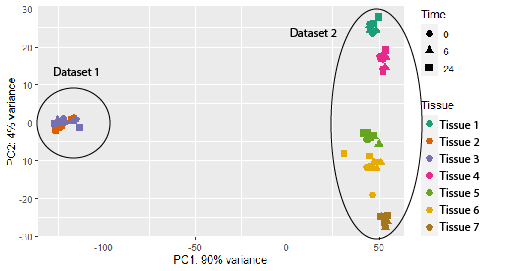
I didn't think it's relevant to add any code or session info as this is a general question.
Thanks in advance! :)

I would like to retrieve a list of target genes in all tissues after certain pathway activation to evaluate to what extent are the target genes shared/different across the tissues. E.g. 0h, 6h and 24h after pathway activation. I can of course still perform this analysis, but just the first dataset comparison after combining the data made me doubt whether there is a better way to normalize the data. Do you think DESeq2 normalization is good enough and I shouldn't worry too much? Thanks!
Given that tissues are nested within dataset, and you are interested in looking at time differences within tissue, I'd recommend processing the two datasets separately, and then comparing at the level of DE gene lists.