
Hi all,
after consulting the manual on data normalization, I have one question left to ask:
The way I see it, there are 4 ways described to obtain normalized data:
The first one is to extract data, normalized using the normalization factors for a gene x sample matrix, and size factors for a single number per sample. This can be done using the following code:
counts(dds, normalized=TRUE)
The second way is to perform log2 transformation log2(n + 1), using the following function:
normTransform(dds)
The third and fourth way is to use the vst and rlog transformation, using the following functions respectively: vst(dds, blind=FALSE) rlog(dds, blind=FALSE)
When I just got started, I used the the first function (counts(dds, normalized=TRUE)), to obtain the normalized data, which I later used for clustering etc. . However, now I doubt that this was the correct decision and that the normalized data, obtained this way, is only used during the DE genes analysis and that for clustering, the second, third and fourth way of normalization is preferred.
I was hoping that any of you could share a more expert opinion on the what normalization to use and whether or not the "counts(dds, normalized=TRUE)" is a viable option as well.
Thank you a lot in advance.
Kind regards, Jonas



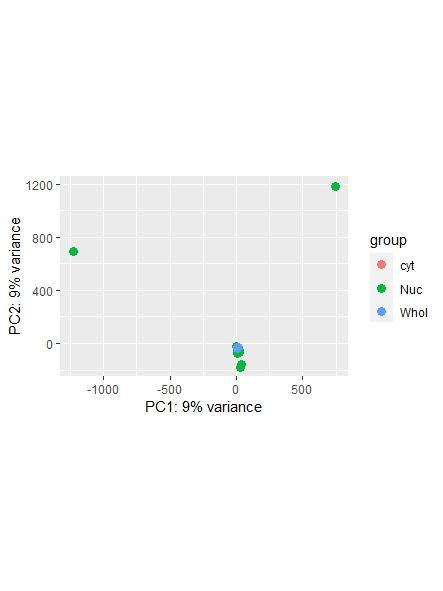
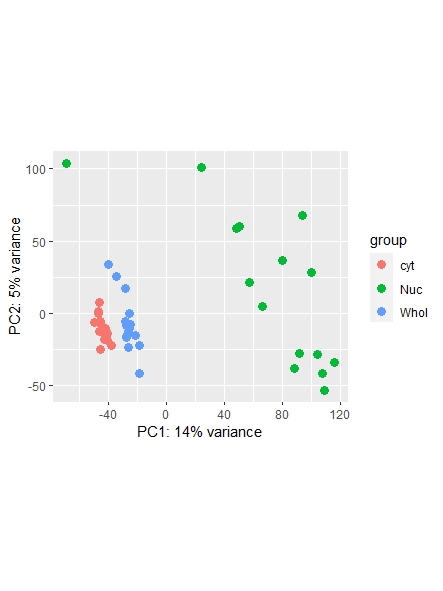
As a side note: I did find a recent question addressing normalization ( https://support.bioconductor.org/p/123651/ ) , however it leaves my question unanswered on whether or not I could also use the counts function ( I guess it's wrong, but I am not sure. Maybe it is still usable... ) and which one is most commonly used/advised. Any opinions shared are much appreciated!
It came to mind that the function: counts(dds, normalized=TRUE), might already return log2 transformed data? (However, this is not described in: https://www.rdocumentation.org/packages/DESeq2/versions/1.12.3/topics/counts)