Entering edit mode

Hi,
I've ~200 samples divided in 7 groups.
After apply DESeq() I wanted to extract the results for several contrasts of interest.
Let say between group A and B.
res <- results(dds,contrast=c("group","B","A"))
resLFC <- lfcShrink(dds, contrast=c("group","B","A"))
Results for res (ordered by p-value) :
log2 fold change (MLE): group B vs A
Wald test p-value: group B vs A
DataFrame with 40705 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000113196.3 38.8525703134052 24.6414633329914 1.7826104729996 13.8232461360598 1.84555955463137e-43 7.46233550319649e-39
ENSG00000110245.12 10.6471323471938 23.3327241057347 2.05828679416885 11.3359927158045 8.70395237188821e-30 1.75967805102464e-25
ENSG00000154122.14 3576.78611413951 1.10788247349132 0.100027172604533 11.0758151474644 1.64377404527271e-28 2.21547865821856e-24
ENSG00000139800.8 13.9621003946128 21.2698946098697 1.96224047614595 10.8395963025113 2.23454907283497e-27 2.25879393027523e-23
ENSG00000288048.1 134.864743190977 4.85765846230169 0.449229418803986 10.8133133293776 2.97718664310579e-27 2.40759129454679e-23
Results for resLFC
log2 fold change (MAP): group B vs A
Wald test p-value: group B vs A
DataFrame with 40705 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue padj
<numeric> <numeric> <numeric> <numeric> <numeric> <numeric>
ENSG00000113196.3 38.8525703134052 -1.27086015761503 0.630634333059615 13.8232461360598 1.84555955463137e-43 7.46233550319649e-39
ENSG00000110245.12 10.6471323471938 0.888268460172144 0.612729047961257 11.3359927158045 8.70395237188821e-30 1.75967805102464e-25
ENSG00000154122.14 3576.78611413951 1.0994369855023 0.0991918038320125 11.0758151474644 1.64377404527271e-28 2.21547865821856e-24
ENSG00000139800.8 13.9621003946128 0.668888551066145 0.619662956340191 10.8395963025113 2.23454907283497e-27 2.25879393027523e-23
ENSG00000288048.1 134.864743190977 4.43259016085307 0.387064306281762 10.8133133293776 2.97718664310579e-27 2.40759129454679e-23
Looking at the log2FC the differences between results() and lfcShrink() are quiet different and even in opposite sign. Which one should I take into account ?
For example the top hit has a log2FC of 24.6 in results() compared to -1.27 in lcfShrink() . Differences is not very good to me. even if p-value are very low. Could someone clarify this point for me ?


Thanks Michael.
I followed your first recommendation :
And here is the differences in terms of adjusted p-values. LRT test seems less conservative. However some of the top hist using LRT are not significant at all in Wald test e.g. ENSG00000159176.14
Ordered by padj_LRT :
Here's the number of genes with padj < 0.01 for both wald and LRT test :
In red : genes with padjLRT < 0.01 and padjwald > 0.01
I'm kind of confuse which test to use. Could you clarify which one to use and/or maybe give me some links explaining when to use either Wald or LRT ?
Thank you Nicolas
I’ve found LRT to be better in these cases of multigroup comparisons with bimodal expression vs 0’s. It is less likely to find these genes to be significant.
Hi Nicolas,
I am just confused if you want to compare the difference among 7 groups, the design should be
design = ~ gender + group, right?Hi Michael,
do yo mean that if I use
test = " LRT", I don't need to shrink?But when I use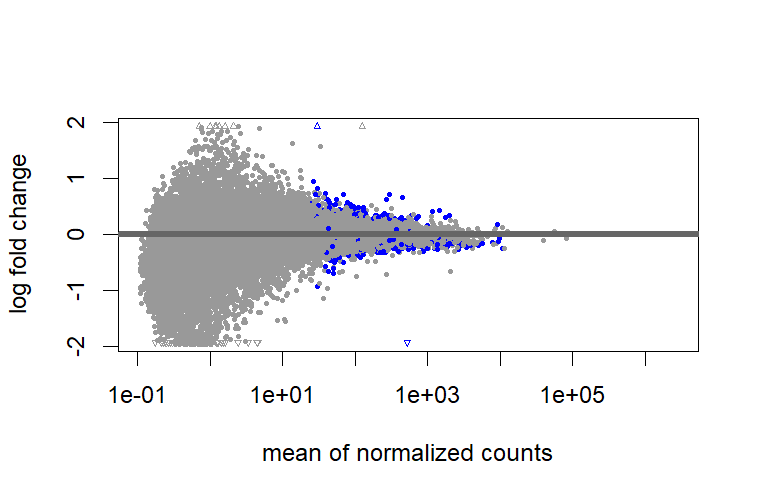
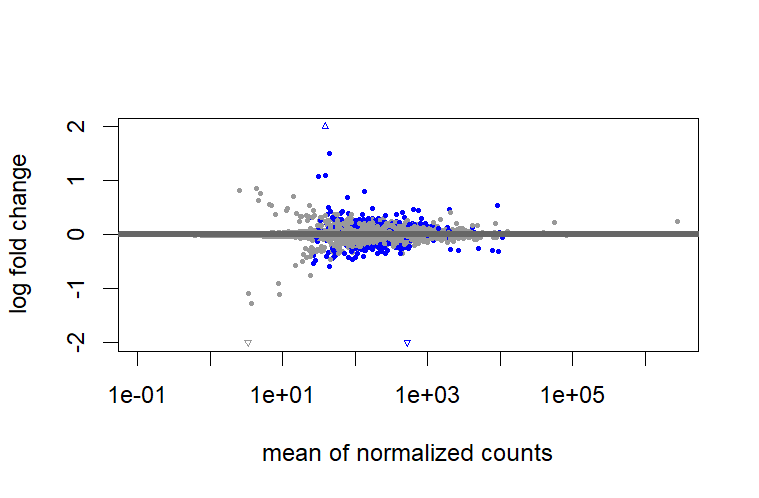
test = " LRT", I found there are big differences between shrunk and unshrunk data. Why and how these differences will influence further analysis?However, it cannot shrink the data for comparison between two groups that are not baseline.
Here, I have Hom, Het and WT 3 groups and WT is the baseline.
It's hard to compare LRT and the posterior estimates. I'd just stick with one approach I think, maybe for you the LRT.
Sorry, could you explain more why we do not need to shrink if using LRT?
LRT gives a p-value which is valid for inference of multiple coefficients. You can just focus on the coefficients from significant genes, as those typically have reliably estimated LFC.