
Hi, I am working on differential expression analysis of multiple leukemia RNA-seq datasets retrieved from SRA. One of my datasets consists of both normal and leukemic samples, whereas the other two are only included leukemic samples. Although I set normal samples as the reference level, the sample distance matrix plot of all datasets clusters samples of one dataset together and samples of other datasets together, no matter they are normal or leukemic. Moreover, the list of significantly expressed genes produced by DESeq2 varies when I use samples of multiple datasets instead of one. I think this problem is rising from different library preparation and sequencing protocol (batch effects) of each dataset if I am right. I would be grateful if someone can help me with fixing this issue to obtain the correct gene list and plot.
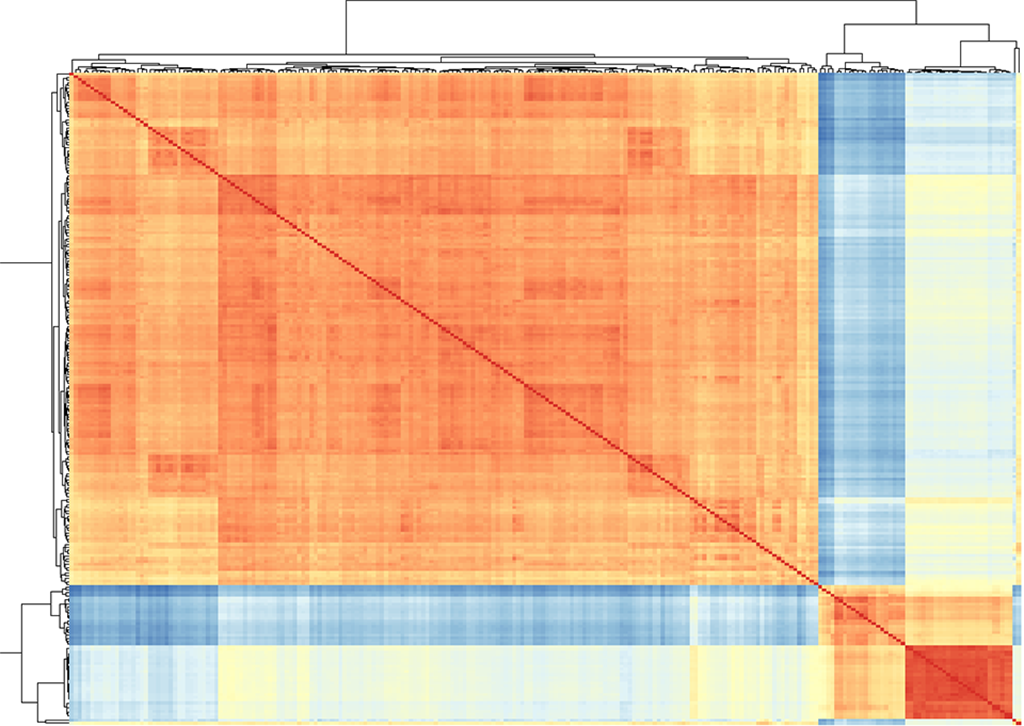

A general comment: Yes, you are combining completely different experiments here, batch effects are almost certainly dominating any biological differences here. I doubt that this can meaningfully be corrected since you only have a single dataset with normals, therefore standard batch correction methods do not apply here. I'd just focus your DE analysis on this dataset. I realize that it is tempting to include more samples to have greater power but in situations like this that does more harm than good. I suggest that for the future (when having non-technical questions that require the developer's expert opinions towards how tools work under the hood) you ask at biostars.org since there is simply a larger user base, and this community here is mainly for technical support of the Bioc packages. There are also plenty of threads on batch correction and the problems that come up when having only one study with both conditions.