
Hi there,
I've just updated Rsamtools to the latest version ("1.22.0") and I've noticed a weird memory leak that is crashing my program.
I have a folder of ~150 bam files that I'm trying to sequentially process, feeding in a data.frame called filter to scanBam(which), that splits the genome into 7744 genomic coordinates.
> head(filter)
V1 V2 V3 V4
1 GL896898 0 30180132 GL896898:0-30180132
2 GL896898 30180133 52375790 GL896898:30180133-52375790
3 GL896899 0 24984016 GL896899:0-24984016
4 GL896899 24984017 40953715 GL896899:24984017-40953715
5 GL896905 0 18438707 GL896905:0-18438707
6 GL896905 18438708 27910907 GL896905:18438708-27910907
For some reason, Rsamtools is not releasing RAM after each iteration. I assumed it was my code, but I just ran the following simple test, and unless I'm being completely stupid, Rsamtools is doing something funny. To test this I looped a single bam file multiple time with the following script and monitored RAM usage:
#R implementation of top for system RAM
library(NCmisc)
#Finds total RAM for all objects stored in Rsession
library(pryr)
library(Rsamtools)
filter=read.table('ferret_merged_sce_events.bed')
objectRam=vector()
totalRam=vector()
for(seq in 1:10)
{
message(paste("iteration", seq))
#Run scanBam to read in bam file
bam.dataframe <- scanBam('bamFile.bam', param=ScanBamParam(which=GRanges(seqnames = c(as.character(filter[,1])), ranges = IRanges(c(filter[,2]), c(filter[,3]) )), mapqFilter=10, what=c("rname","pos","strand")))
#measure RAM across all R objects
objectRam <- c(objectRam, mem_used()[1]/10^6)
#measure RAM across system
totalRam <- c(totalRam, suppressWarnings(top(CPU=F)$RAM$used*10^6))
#remove scanBam object to reset for next iteration
rm(bam.dataframe)
#empty garbage collection
gc()
}
plot(objectRam, type='l', xlab='iteration', ylab='Ram usage (Mb)')
plot(totalRam, type='l', xlab='iteration', ylab='Ram usage (Mb)')
This generates the two attached graphs. You'll see that while the RAM used for the R object remains stable, the overall RAM usage of my computer increases, and only decreases when I end my R session. Is there any way of purging the RAM? Is this a bug in Rsamtools or have I done something wrong?
The amount of memory used for all the objects in my R session is constant:
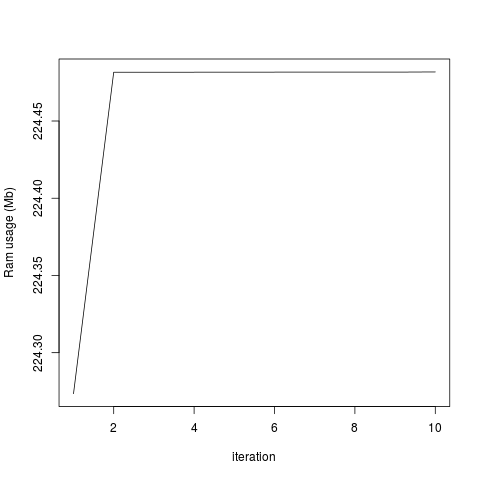
...But the total system usage of RAM increases with each iteration:
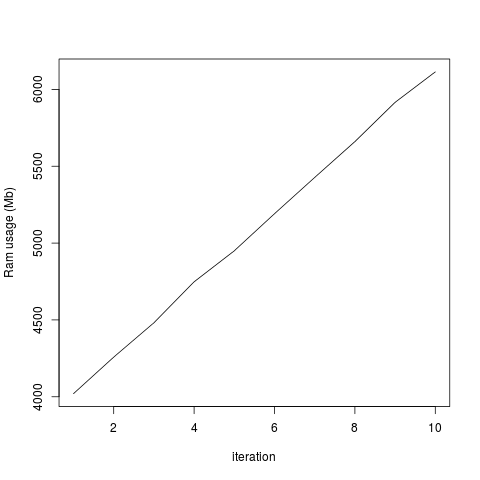
I don't think this happened in earlier versions of Rsamtools, as I'd previously analyzed these data without a problem. I added the rm() and gc() lines in the hope it would fix the problem, but it doesn't seem to change anything. I tried running this without the which= option in scanBamParam() and there doesn't seem to be this memory retention issue, so it's specific for scanBam when which is specified as a parameter. Any help would be greatly appreciated!
Just to be clear; this doesn't happen with BioC 3.1, right? I'm asking because I'm also trying to diagnose a BioC 3.2-specific problem in some code that relies heavily on
scanBam(and thewhich=option). It's proving quite difficult to pin down the cause, which would be consistent with memory problems.Hi Aaron,
I'm not sure; I updated Bioconductor and Rsamtools at the same time. I'm on Bioconductor v3.2 on R v3.2.2 using Rsamtools v1.22.0. My code was working fine on Bioconductor v3.1 with Rsamtools v1.18, but after updating it poops out after it loops through ~40 samples with a memory usage error. I haven't installed earlier versions yet because I really wanted the mapqFilter option, which wasn't available in Rsamtools v1.18 but massively speeds up my code since I don't need to convert to the result of scanBam() to a data.frame to filter. I wrote the simple loop script to convince myself that the problem wasn't my code, but something strange happening with the Rsamtools function.
Hmm... your problem might be related to mine. For anyone who's interested, my issue is that the ChIP-seq DB workflow runs extremely slowly in BioC 3.2, while being fine in BioC 3.1. Specifically, the second set of
aligncalls fromRsubreadtakes a very long time (~18 hours per file, which is close to a 5- to 10-fold slowdown compared to BioC 3.1). Running thosealigncalls separately doesn't result in any problems; it's only when they're in the workflow (with lots of preceding calls toscanBamby variouscsawfunctions) that this drop in speed occurs. My uneducated guess is that, if memory is not being released byRsamtools, thenRsubreadis slowing down to operate in what it perceives to be a low-memory environment. Oh - this probably doesn't help you too much, though; apologies for going off-topic.Could well be related. Have you monitored RAM? Our lab is pretty small fry, so I'm only on a computer with 16Gb RAM to do all the analysis. After getting the inital error, I monitored with a separate terminal running top -p `pgrep R | tr "\\n" "," | sed 's/,$//' command, and just watched my memory slowly sucked away. No problem about going off topic; it sounds like this could be related. I guess if you saved the product of Rsamtools, then restarted R, called the saved object and continued you could see if your downstream workflow functions are still slow.
RAM doesn't seem to be the problem; I'm running the workflow on a server with plenty of memory, and usage is around 10G's, which is a small proportion of the total. Rather, the concern has more to do with whether the memory problem is interfering with allocations later on, i.e., in
Rsubread. An additional distinction between our problems is that theRsubreadallocations are done usingmalloc/freeinstead of going through R's API.Can you simplify your example a little, by creating the ScanBamParam once, outside the loop, and not saving the result to bam.dataframe? Is it possible to reproduce the phenomenon on publicly available BAM files, e.g., in RNAseqData.HNRNPC.bam.chr14 ? (an emerging pet peeve --
message("foo ", "bar")is the same asmessage(paste("foo", "bar"));).