Entering edit mode

Hi, Dr Dr Love
I find a werid MAplot or volcano plot of DESeq reuslt. I am wondering whether you can give me some advice.
This diff result is from two cell type bulk RNA-seq. I use two specific marker to get these two cell type using Flow cytometer. I alreadly know these cell type have very different RNA composition.
As you can see, the MA-plot is not like DESeq2 manul example. And the volcano plot is more werid, it looks like -log10(padj) is the linear function of log2FoldChange.
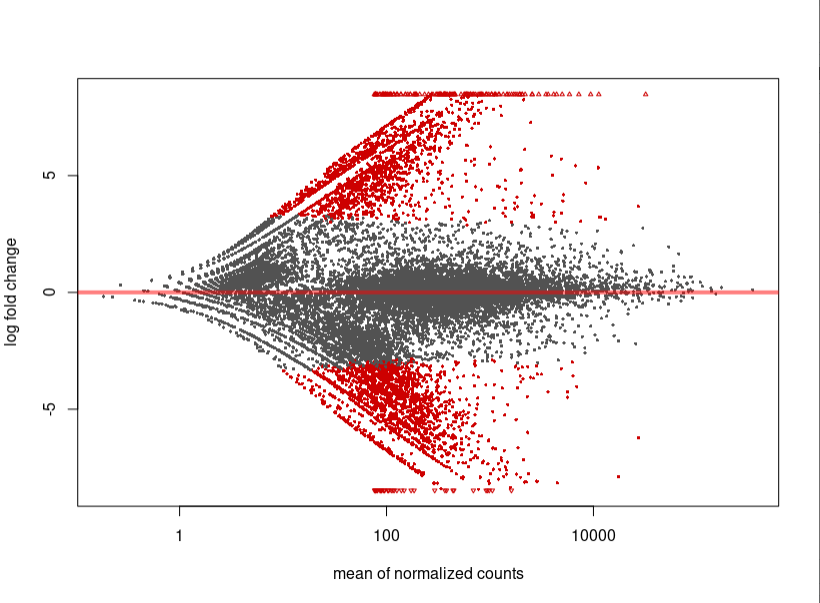
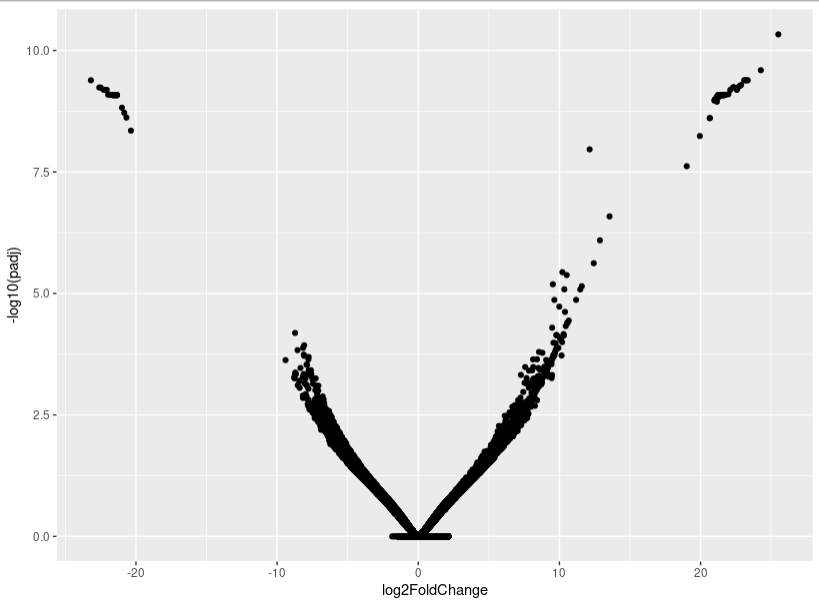
If you want to repeat my data, here is my data link:
https://github.com/shangguandong1996/picture_link/blob/main/WFX_count_Rmatrix.txt
And here is my code
# Prepare -----------------------------------------------------------------
# load up the packages
library(DESeq2)
library(dplyr)
library(ggplot2)
# Set Options
options(stringsAsFactors = F)
# load up the data
data <- read.table("rawdata/count/WFX_count_Rmatrix.txt",
header = TRUE,
row.names = 1)
coldata <- data.frame(row.names = colnames(data),
type = rep(c("Fx593", "Fx600"), each = 2))
# DESeq2 ------------------------------------------------------------------
dds <- DESeqDataSetFromMatrix(countData = data,
colData = coldata,
design= ~ type)
# PCA
vsd <- vst(dds)
plotPCA(vsd, intgroup = "type")
dds <- DESeq(dds)
res_lfc <- lfcShrink(dds = dds,
type = "ashr",
coef = "type_Fx600_vs_Fx593")
plotMA(res_lfc)
as_tibble(res_lfc) %>%
mutate(padj = case_when(
is.na(padj) ~ 1,
TRUE ~ padj
)) %>%
ggplot(aes(x = log2FoldChange, y = -log10(padj))) +
geom_point()
> sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /opt/sysoft/R-3.6.1/lib64/R/lib/libRblas.so
LAPACK: /opt/sysoft/R-3.6.1/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods
[9] base
other attached packages:
[1] ggplot2_3.3.3 dplyr_1.0.5
[3] DESeq2_1.26.0 SummarizedExperiment_1.16.0
[5] DelayedArray_0.12.0 BiocParallel_1.19.6
[7] matrixStats_0.58.0 Biobase_2.46.0
[9] GenomicRanges_1.38.0 GenomeInfoDb_1.22.0
[11] IRanges_2.20.0 S4Vectors_0.24.0
[13] BiocGenerics_0.32.0
loaded via a namespace (and not attached):
[1] bitops_1.0-7 bit64_4.0.5 doParallel_1.0.15
[4] RColorBrewer_1.1-2 tools_3.6.1 backports_1.2.1
[7] utf8_1.2.1 R6_2.5.0 rpart_4.1-15
[10] Hmisc_4.5-0 DBI_1.1.1 colorspace_2.0-1
[13] nnet_7.3-16 withr_2.4.2 tidyselect_1.1.0
[16] gridExtra_2.3 bit_4.0.4 compiler_3.6.1
[19] cli_2.5.0 htmlTable_2.1.0 labeling_0.4.2
[22] scales_1.1.1 checkmate_2.0.0 SQUAREM_2017.10-1
[25] genefilter_1.68.0 mixsqp_0.2-2 stringr_1.4.0
[28] digest_0.6.27 foreign_0.8-73 XVector_0.26.0
[31] pscl_1.5.2 base64enc_0.1-3 jpeg_0.1-8.1
[34] pkgconfig_2.0.3 htmltools_0.5.1.1 fastmap_1.1.0
[37] htmlwidgets_1.5.3 rlang_0.4.11 rstudioapi_0.13
[40] RSQLite_2.2.7 generics_0.1.0 farver_2.1.0
[43] RCurl_1.98-1.3 magrittr_2.0.1 GenomeInfoDbData_1.2.2
[46] Formula_1.2-4 Matrix_1.3-2 Rcpp_1.0.6
[49] munsell_0.5.0 fansi_0.4.2 lifecycle_1.0.0
[52] stringi_1.5.3 MASS_7.3-54 zlibbioc_1.32.0
[55] grid_3.6.1 blob_1.2.1 crayon_1.4.1
[58] lattice_0.20-44 splines_3.6.1 annotate_1.64.0
[61] locfit_1.5-9.4 knitr_1.33 pillar_1.6.0
[64] geneplotter_1.64.0 codetools_0.2-18 XML_3.99-0.3
[67] glue_1.4.1 packrat_0.5.0 latticeExtra_0.6-29
[70] data.table_1.12.6 png_0.1-7 vctrs_0.3.8
[73] foreach_1.4.7 gtable_0.3.0 purrr_0.3.3
[76] assertthat_0.2.1 ashr_2.2-39 cachem_1.0.4
[79] xfun_0.22 xtable_1.8-4 survival_3.2-11
[82] truncnorm_1.0-8 tibble_3.1.1 iterators_1.0.12
[85] AnnotationDbi_1.48.0 memoise_2.0.0 cluster_2.1.2
[88] ellipsis_0.3.2

This is probably a consequence of many rows with overall very low counts.
Here you see that only about 1/3 of your rows actually have more than 10 raw counts in at least two of the four samples. I guess this is where some pre-filtering will help removing spurious calls.
Thanks, AT. After
The MA or volcano plot is better. But I am wondering whether there are other advices for this data type, after all, this data type is "pseudo single-cell RNA-seq".
I also post a question in biostar
https://www.biostars.org/p/9484986/