Entering edit mode

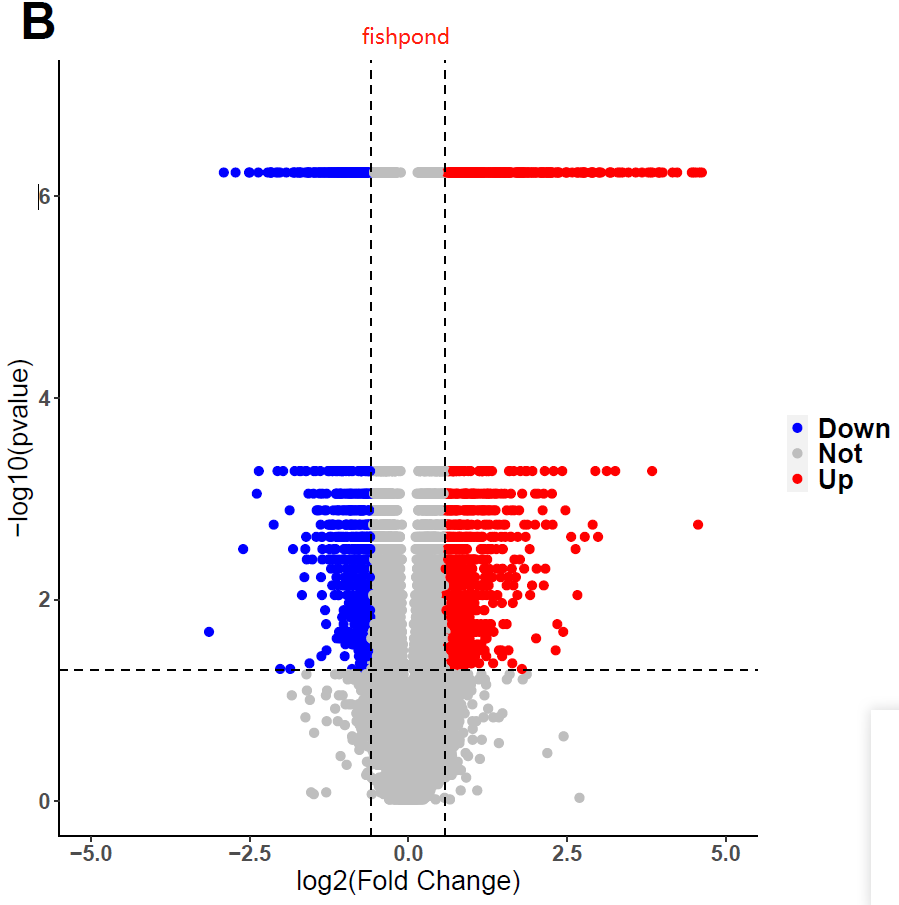
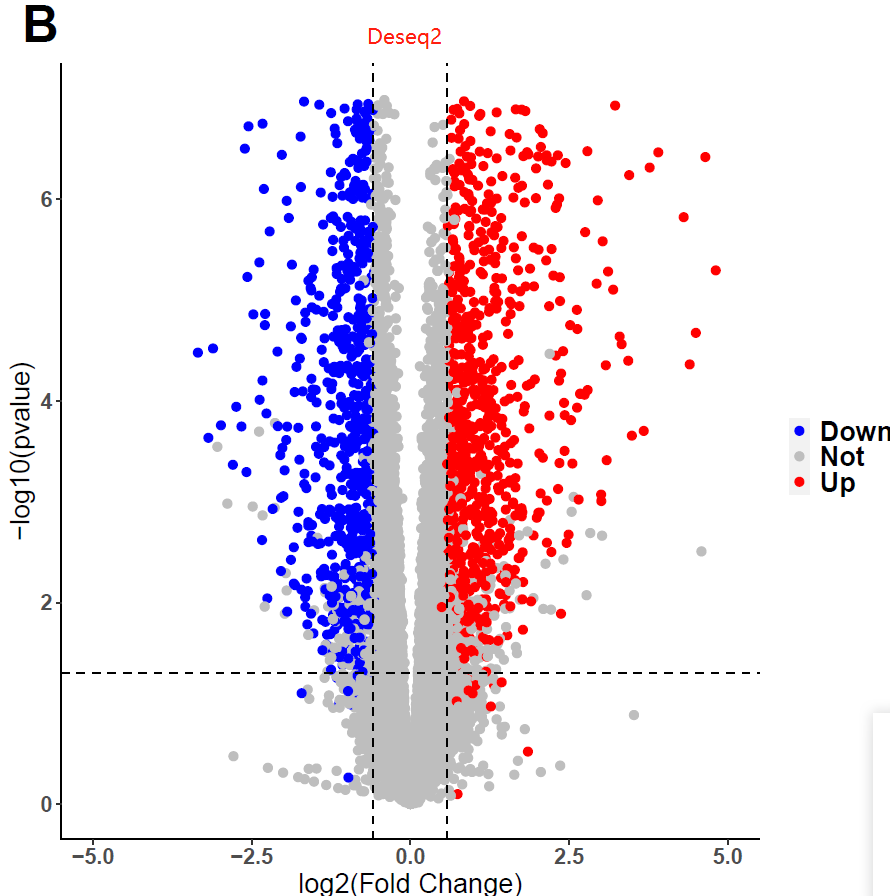
I used fishpond for DEGs analysis, but I obtained weird pvalue. I tried many datasets and parameters, still in vain. Interestingly, I found I would get conventional distribution of pvalues, if i used DESeq2 instead of fishpond. Does fishpond lead these? Are the results of fishpond Credible? Or, did I select unfit parameters? If you want the codes, I can send to you!
Best, Seager
#1 Part of quant's codes
######################################
if [[ $LIB -eq 2 ]]; then # Check whether the library is paired
salmon quant \
-i $indexdir \
-l A \
-1 $workdir/fastq/$line".read1.fq" \
-2 $workdir/fastq/$line".read2.fq" \
-p 44 \
-o $workdir/salmon_out/quant_out/$line \
-g $gtfpath \
--numGibbsSamples 5 \
--auxDir aux_info \
--seqBias --gcBias -d --posBias --hardFilter --discardOrphansQuasi --writeUnmappedNames && echo salmon $line Done!
# note: rabbitQC(ktrim) outputs with suffix, "read1.fa" and "read2.fq"; change "--dumpEq" to "-d";add "--hardFilter" and "--discardOrphansQuasi"; "-l ISR" to "-l A"
date
else
salmon quant \
-i $indexdir \
-l A \
-r $workdir/fastq/$line".read.fq" \
-p 44 \
-o $workdir/salmon_out/quant_out/$line \
-g $gtfpath \
--numGibbsSamples 5 \
--auxDir aux_info \
--seqBias --gcBias -d --posBias --hardFilter --discardOrphansQuasi --writeUnmappedNames && echo salmon $line Done!
# note: rabbitQC(ktrim) outputs with suffix, "read1.fa" and "read2.fq"; change "--dumpEq" to "-d";add "--hardFilter" and "--discardOrphansQuasi"; "-l ISR" to "-l A"
date
fi
######################################
#2 codes of fishpond
library(tximeta)
library(fishpond)
library(data.table)
library(tidytable)
library(DESeq2)
suppressPackageStartupMessages(library(SummarizedExperiment))
dir<- "/home2/ymwang/linqin/RNA-seq/Analysis/GSE112055_t2/salmon_out/"
pdata <- fread("dataset_TAC.txt", sep="\t", header=T)
##1 coldata <- pdata %>% drop_na.(Day) %>% filter.(`Series ID` == "GSE66630" | `Series ID` == "GSE112055") ## Just use RNA-seq samples with days and GSE66630&GSE112055
coldata <- pdata %>% drop_na.(Day) %>% filter.(`Series ID` == "GSE112055") ## Just use RNA-seq samples with days and GSE112055
colnames(coldata)[grep("Run$",colnames(coldata))] <-"names" ## Note: fishpond need colnames "names"
head(coldata)
coldata$files <- file.path(dir, "quant_out",coldata$names, "quant.sf")
all(file.exists(coldata$files))
head(coldata)
makeLinkedTxome(
indexDir = "/home2/ymwang/linqin/RNA-seq/Genome/release102/mouse/index_mouse_r102_gffread_salmon_k31",
source = "Ensembl",
organism = "Mus musculus",
release = "102",
genome = "GRChm38",
fasta = "/home2/ymwang/linqin/RNA-seq/Genome/release102/mouse/transcript_gffread.fa",
gtf = "/home2/ymwang/linqin/RNA-seq/Genome/release102/mouse/Mus_musculus.GRCm38.102.gtf",
write = FALSE
)
se <- tximeta(coldata) # delete skipMeta=TRUE
assayNames(se)
head(rownames(se))
se
class(se)
cat("tximeta OK!")
##2 differential expression analysis at gene level
gse <- summarizeToGene(se)
#2.1 fishpond
y <- gse[,gse$Condition %in% c("Sham-w5", "TAC-w5")]
y$Condition <- factor(y$Condition, c("Sham-w5", "TAC-w5")) #the ordering of 'Sham' and 'TAC' will determine the log2FC(here is log2(TAC/Sham))
y
y <- scaleInfReps(y)
y <- labelKeep(y)
y <- y[mcols(y)$keep,]
cat("scaleInfReps-labelKeep OK!")
set.seed(910)
out <- swish(y, x="Condition", nperms=100)
out
head(mcols(out))
cat("swish OK!")
res1 <- as_tidytable(mcols(out))
# fwrite(res1,'DEGs_gse112055_fishpond.txt',sep="\t",quote=F)
cat("fishpond OK!")
#2.2 deseq2
dds <- DESeqDataSet(y, design = ~ Condition) # y getting from above
dds <- DESeq(dds)
dds$Condition <- factor(dds$Condition, levels =c("Sham-w5", "TAC-w5"))
res2 <- results(dds)
res2
##3 plot
library(data.table)
library(tidytable)
library(ggplot2)
require(grid)
library(Rmisc)
## fishpond
data <- res1
threshold <- as.factor(ifelse(data$pvalue <= 0.05 & abs(data$log2FC) >= log2(1.5) , ifelse(data$log2FC >= log2(1.5) ,'Up','Down'),'Not'))
pdf("plot1.pdf")
ggplot(data,aes(x=log2FC,y=-log10(pvalue),colour=threshold)) +
xlab("log2(Fold Change)")+ylab("-log10(pvalue)") +
geom_point(size = 2,alpha=1) +
ylim(0,7) + xlim(-5,5) +
scale_color_manual(values=c("blue","grey", "red"))+
geom_vline(xintercept = c(-log2(1.5), log2(1.5)), lty = 2,colour="#000000")+
geom_hline(yintercept = c(-log10(0.05)), lty = 2,colour="#000000") +
ggtitle("B") +
guides(fill = guide_legend(reverse = F))+
theme(plot.title = element_text(hjust = -0.06,size = 28, face = "bold"),
legend.title = element_blank(),
legend.text = element_text(size = 15, face = "bold"),
legend.position = 'right',
legend.key.size=unit(0.4,'cm'))+
theme(panel.grid.major =element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"),axis.text=element_text(size=12,face = "bold"),axis.title.x=element_text(size=15),axis.title.y=element_text(size=15))
dev.off()
# deseq2, note: log2FoldChange in deseq2
data <- data.frame(res2)
pdf("plot2.pdf")
ggplot(data,aes(x=log2FoldChange,y=-log10(pvalue),colour=threshold)) +
xlab("log2(Fold Change)")+ylab("-log10(pvalue)") +
geom_point(size = 2,alpha=1) +
ylim(0,7) + xlim(-5,5) +
scale_color_manual(values=c("blue","grey", "red"))+
geom_vline(xintercept = c(-log2(1.5), log2(1.5)), lty = 2,colour="#000000")+
geom_hline(yintercept = c(-log10(0.05)), lty = 2,colour="#000000") +
ggtitle("B") +
guides(fill = guide_legend(reverse = F))+
theme(plot.title = element_text(hjust = -0.06,size = 28, face = "bold"),
legend.title = element_blank(),
legend.text = element_text(size = 15, face = "bold"),
legend.position = 'right',
legend.key.size=unit(0.4,'cm'))+
theme(panel.grid.major =element_blank(), panel.grid.minor = element_blank(),panel.background = element_blank(), axis.line = element_line(colour = "black"),axis.text=element_text(size=12,face = "bold"),axis.title.x=element_text(size=15),axis.title.y=element_text(size=15))
dev.off()
print("All done!")
sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-conda_cos6-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /home2/ymwang/miniconda3/envs/R40/lib/libopenblasp-r0.3.12.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] Rmisc_1.5 plyr_1.8.6
[3] lattice_0.20-44 ggplot2_3.3.5
[5] DESeq2_1.30.1 SummarizedExperiment_1.20.0
[7] Biobase_2.50.0 MatrixGenerics_1.2.1
[9] matrixStats_0.60.1 GenomicRanges_1.42.0
[11] GenomeInfoDb_1.26.7 IRanges_2.24.1
[13] S4Vectors_0.28.1 BiocGenerics_0.36.1
[15] tidytable_0.6.5 data.table_1.14.0
[17] fishpond_1.6.0 tximeta_1.9.5
loaded via a namespace (and not attached):
[1] colorspace_2.0-2 ellipsis_0.3.2
[3] qvalue_2.22.0 XVector_0.30.0
[5] rstudioapi_0.13 farver_2.1.0
[7] bit64_4.0.5 interactiveDisplayBase_1.28.0
[9] AnnotationDbi_1.52.0 fansi_0.5.0
[11] xml2_1.3.2 splines_4.0.2
[13] tximport_1.18.0 cachem_1.0.6
[15] geneplotter_1.68.0 jsonlite_1.7.2
[17] Rsamtools_2.6.0 annotate_1.68.0
[19] dbplyr_2.1.1 shiny_1.6.0
[21] BiocManager_1.30.16 readr_2.0.1
[23] compiler_4.0.2 httr_1.4.2
[25] assertthat_0.2.1 Matrix_1.3-4
[27] fastmap_1.1.0 lazyeval_0.2.2
[29] cli_3.0.1 later_1.3.0
[31] htmltools_0.5.2 prettyunits_1.1.1
[33] tools_4.0.2 gtable_0.3.0
[35] glue_1.4.2 GenomeInfoDbData_1.2.4
[37] reshape2_1.4.4 dplyr_1.0.7
[39] rappdirs_0.3.3 Rcpp_1.0.7
[41] vctrs_0.3.8 Biostrings_2.58.0
[43] rtracklayer_1.50.0 stringr_1.4.0
[45] mime_0.11 lifecycle_1.0.0
[47] ensembldb_2.14.1 gtools_3.9.2
[49] XML_3.99-0.7 AnnotationHub_2.22.1
[51] zlibbioc_1.36.0 scales_1.1.1
[53] vroom_1.5.4 hms_1.1.0
[55] promises_1.2.0.1 ProtGenerics_1.22.0
[57] AnnotationFilter_1.14.0 RColorBrewer_1.1-2
[59] yaml_2.2.1 curl_4.3.2
[61] memoise_2.0.0 biomaRt_2.46.3
[63] stringi_1.7.4 RSQLite_2.2.8
[65] BiocVersion_3.12.0 genefilter_1.72.1
[67] GenomicFeatures_1.42.3 BiocParallel_1.24.1
[69] rlang_0.4.11 pkgconfig_2.0.3
[71] bitops_1.0-7 purrr_0.3.4
[73] labeling_0.4.2 GenomicAlignments_1.26.0
[75] bit_4.0.4 tidyselect_1.1.1
[77] magrittr_2.0.1 R6_2.5.1
[79] generics_0.1.0 DelayedArray_0.16.3
[81] DBI_1.1.1 pillar_1.6.2
[83] withr_2.4.2 survival_3.2-13
[85] abind_1.4-5 RCurl_1.98-1.4
[87] tibble_3.1.4 crayon_1.4.1
[89] utf8_1.2.2 BiocFileCache_1.14.0
[91] tzdb_0.1.2 progress_1.2.2
[93] locfit_1.5-9.4 blob_1.2.2
[95] digest_0.6.27 xtable_1.8-4
[97] httpuv_1.6.3 openssl_1.4.5
[99] munsell_0.5.0 askpass_1.1

Thans for your reply. I'm sorry for reply so late, because I didn't getting any messages in my email. I will read details of the vignette.