
Hello!
I am analyzing my RNA-seq data using the DESeq2 method. I am using the following code to plot the 50 most differentially expressed genes in my database, but in my biological triplicate, I have one sample that acts as an outlier in the expression of some genes but looks congruent with the expression of the other replicates when I consider a large number of genes. !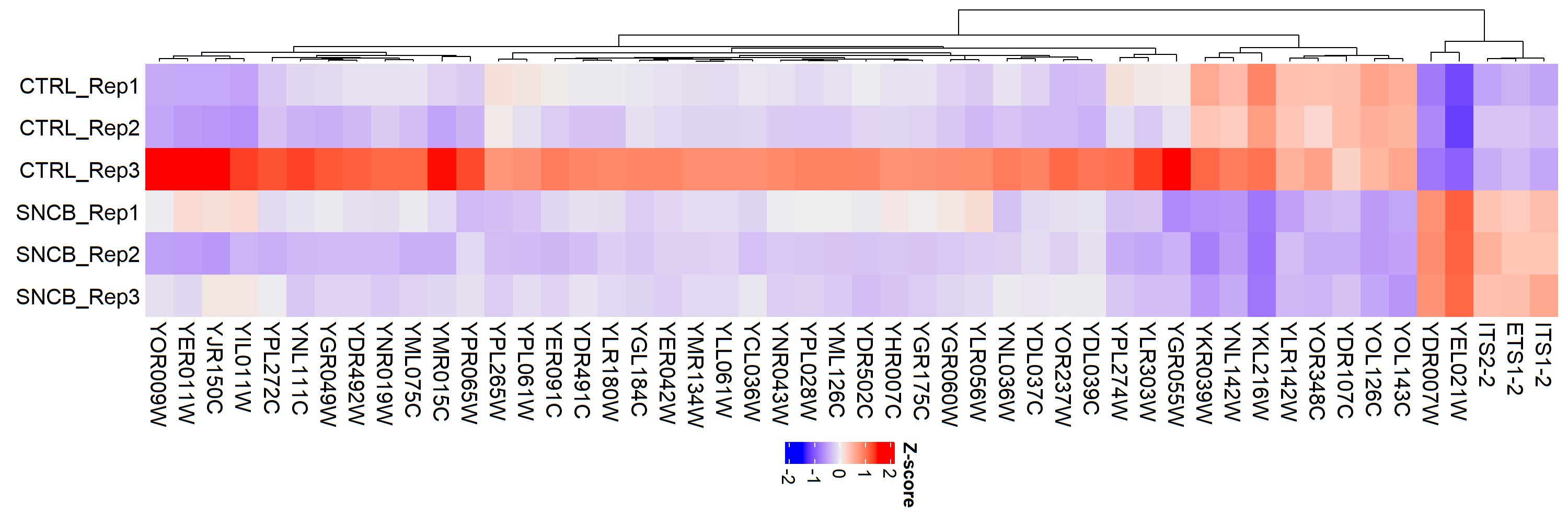
I apologize if my question is not correct or if I am not using the proper words, I started recently to use DESeq2 and I am not so proficient in coding, so I am a beginner in the RNAseq analysis.
Thank you for your time and your help.
Code should be placed in three backticks as shown below
```# select the 50 most differentially expressed genes topVarGenes <- head(order(rowVars(assay(rld)), decreasing = TRUE), 50)
mat <- assay(rld)[ topVarGenes, ] mat <- mat - rowMeans(mat) anno <- as.data.frame(mat)
library(ComplexHeatmap) library(ggplot2) f <- Heatmap(mat, cluster_rows = T, cluster_columns = F, column_labels = colnames(anno), name = "Z-score") png('Heatmap_1oo.png', res = 250, width = 1000, height = 3000) print(f) dev.off()
```
