
Hi,
I have two conceptual/technical questions about spike-in normalisation of ChIP-seq data.
We did spike-in for PolII ChIP-seq from a mouse cell line with ActiveMotif kit, which uses anti-H2Av antibody with drosophila cells. The mouse and fly chromatin are mixed and a mix of anti-mouse PolII and anti-H2Av antibodies are used to IP. I aligned the reads to mouse and drosophila genome separately. And now I am trying to get normalization factors for each replicate using DiffBind (v. 3.8.3), which I would like to use for differential peak analysis as well as scaling bigwig files.
Qn1: From what I can gather, in DiffBind, by including the spikein bam filles in the Spikein column of sampleSheet, I have the option to use the spikein library size or the csaw background bins method with RLE or TMM. Is there a built-in option to use the reads in peaks (RiP) method from the spike-in datasets (i.e., read counts in H2Av peaks) for normalizing the mouse reads? If there is an option to supply the peaks in the spike-in dataset, I cannot find it.
To my naive thinking, this option better controls for IP efficiency (~signal/noise ratio) per replicate than library size or background bins (assuming the antibody and chromatin concentrations in the replicates are constant). Am I missing something conceptually?
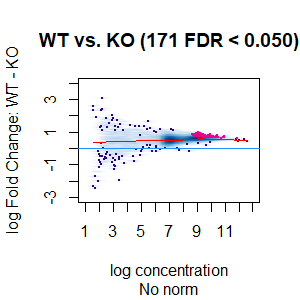
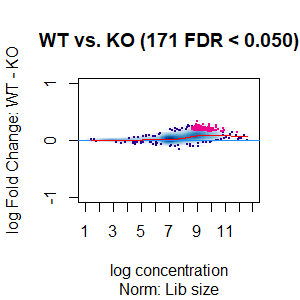
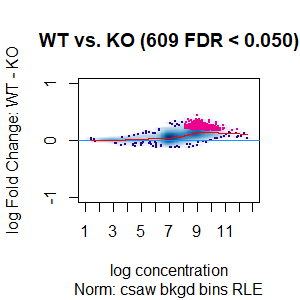
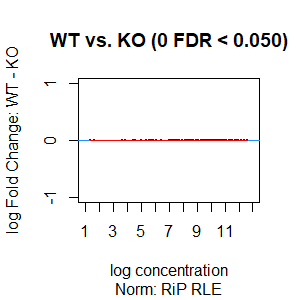
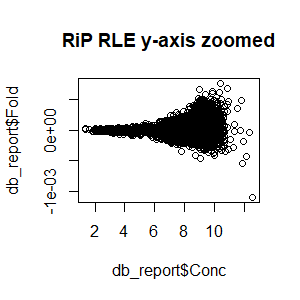
Qn2: To get the normalisation factors using the RiP from the spike-in datasets, I made a dba object using the drosophila sequences ONLY (drosophila bam files + H2Av peaks) and tried the various normalisations (code below). Differential peaks should not be found between conditions here, since these are spike-in chromatin IPs. It looks like the RiP RLE normalisation does an eerily excellent job at normalising the counts, while the library size or background bins methods still give a lot significantly different peaks when they shouldn't (although the fold change is minimal).
I understand, with RiP RLE normalisation, I am more directly normalising the peak signals and plotting the peak signal, so the fact that it has the best effect in this analysis is not surprising in itself. But I am surprised by the near absence of variation between the replicates (meaning a lot of the variation comes from IP efficiency between samples?). Does this mean, RiP RLE normalisation using spike-in peaks is the correct method to normalise my mouse read counts?
I am pretty new to ChIP-seq/DiffBind analyses. Please let me know if anything is wrong or unclear. Thank you!
>spik.db <- dba(sampleSheet = "../input_data/sampleSheets/P2_dm3_spikein_sampleSheet_DiffBind.csv",
minOverlap = 1)
>spik.db
4 Samples, 11449 sites in matrix:
ID Tissue Factor Condition Treatment Replicate Intervals
1 1 CL P2 WT Full-media 1 10715
2 2 CL P2 WT Full-media 2 9763
3 3 CL P2 KO Full-media 1 11166
4 4 CL P2 KO Full-media 2 11480
>spik.counts <- dba.count(spik.db,minOverlap = 1)
>spik.model <- dba.contrast(spik.counts, minMembers=2, reorderMeta=list(Condition="KO"))
>spik.model <- dba.analyze(spik.model) #default lib size
>dba.plotMA(spik.model, bNormalized = F, sub="No norm")
>dba.plotMA(spik.model, bNormalized = T, sub="Norm: Lib size")
>rm(spik.model)
>spik.model <- dba.contrast(spik.counts, minMembers=2, reorderMeta=list(Condition="KO"))
>spik.model<-dba.normalize(spik.model,normalize = "RLE",background=T) # bkgd bins RLE normalisation
>spik.model <- dba.analyze(spik.model)
>dba.plotMA(spik.model, bNormalized = T, sub="Norm: csaw bkgd bins RLE")
>rm(spik.model)
>spik.model <- dba.contrast(spik.counts, minMembers=2, reorderMeta=list(Condition="KO"))
>spik.model<-dba.normalize(spik.model,normalize = "RLE") # RiP RLE normalisation
>spik.model <- dba.analyze(spik.model)
>dba.plotMA(spik.model, bNormalized = T, sub="Norm: RiP RLE")
>db_report<-dba.report(spik.model,th=1,bCounts = T) #to examine normalized counts of RiP RLE
>plot(db_report$Fold~db_report$Conc,main="RLE (RiP) y-axis zoomed")

I looked at the
dba.normalize()help section where aGRangescan be supplied to thespikeinparameter. This is the same interface for supplying parallel factor normalisation right?I have two chromosomes that have the same names in mouse and drosophila genomes. Would that be a problem? It is a little bit unclear to me, whether supplying the spike-in bam files in the sample sheet forces
dba.normalize()to look at theGRangesin itsspikeinparameter, only ineg.db.obj$samples$Spikeinbam files or alsoeg.db.obj$samples$bamReads. I didn't realise this would be an issue downstream when aligning, or I would have aligned to the combined genomes with the fly chromosome names altered (probably best practice going forward regardless).I would also like to edit the original question to reflect that TMM normalisation produces almost identical results to RLE. The distinction is really whether reads in peaks (RiP) vs library size/background bins method for this specific use case. But, I do not know the etiquette for editing after the question has been replied to. If it is frowned upon, this comment can be the record clarifying this.
And thanks a lot for actively supporting the package, responding to questions and the tutorials Rory. I learnt a lot about DiffBind from your replies to the different forum posts!
Yes, this is the same interface as for parallel factors. The difference is whether or not
Spikeinfiles are specified in the sample sheet. If there are noSpikeinfiles supplied, theGRangeswill be evaluated against the primary read count files, as in the case of a parallel factor. When you specifySpikeinfiles, only those files will be looked at, so in your case where they share a chromosome name, only reads on that chromosome in theSpikeinfiles will be counted.Yes, I can confirm supplying spikein peaks this way works as you mentioned.
After looking at the RiP proportions for mouse PolII and Drosophila H2AV in their respective genomes, I do have a follow-up conceptual question. The RiP values are not consistently changing between the replicates (i.e., one replicate has less PolII RIP and more H2AV RiP and the other replicate is the opposite). Assuming RiP proportion reflects IP efficiency, the IP efficiencies between PolII and H2AV antibodies do not seem to change consistently, meaning I cannot confidently use H2AV IP efficiency to normalise PolII IP. I guess if I had used a single antibody (a PolII antibody that binds to both mouse and Drosophila PolII), then mouse and Drosophila RiP values will more consistently change between replicates? Have you come across this scenario? Am I overthinking about this inconsistency mouse/Drosophila RiP proportion between replicates of the same sample? In the end, I am using the csaw background bins method with RLE on the spikein H2AV reads to normalise mouse PolII reads.