Entering edit mode

morteza.aslanzadeh
▴
30
@mortezaaslanzadeh-23584
Last seen 4.6 years ago
I'm using TopGo to do GO analyses. When I get my enriched terms, the long term names get corrupted. I have some text then .... For example:
GO:0000122 negative regulation of transcription by ...
GO:0006886 intracellular protein transport
GO:0010976 positive regulation of neuron projection...
Any help would be appreciated?
My entire code:
library(topGO)
library(GO.db)
library(biomaRt)
library(Rgraphviz)
library(tidyverse)
library(dbplyr)
prebg_genes <- read.table("mart_mmu_protein_coding_genes.csv", header=F, sep=",")
bg_genes <- as.character(prebg_genes[,1])
pre_candidate_list_1 <- read.table("All_targets_ENSMUSG.txt", header=T, sep=",")
candidate_list_1= as.character(pre_candidate_list_1[,1])
db <- useMart('ENSEMBL_MART_ENSEMBL',dataset='mmusculus_gene_ensembl', host="www.ensembl.org")
go_ids <- getBM(attributes=c('go_id', 'ensembl_gene_id', 'namespace_1003'), filters='ensembl_gene_id', values=bg_genes, mart=db)
go_ids <- read.table("go_ids_mmu_protein_coding_genes.txt", header=T, sep="\t")
listAttributes(db)
View(go_ids)
gene_2_GO <- unstack(go_ids[,c(1,2)])
pre_keep1 <- candidate_list_1 %in% go_ids[,2]
keep1 <- which(pre_keep1==TRUE)
candidate_list_final1 <- candidate_list_1[keep1]
geneList1 <- factor(as.integer(bg_genes %in% candidate_list_final1))
names(geneList1) <- bg_genes
GOdata1 <- new('topGOdata', ontology='BP', allGenes = geneList1, annot = annFUN.gene2GO, gene2GO = gene_2_GO)
classic_fisher_result1 <- runTest(GOdata1, algorithm='classic', statistic='fisher')
weight_fisher_result1 <- runTest(GOdata1, algorithm='weight01', statistic='fisher')
applied to the topGOdata object.
allGO1 <- usedGO(GOdata1)
all_res1 <- GenTable(GOdata1, weightFisher=weight_fisher_result1, orderBy='weightFisher', topNodes=length(allGO1))
p.adj1 <- round(p.adjust(all_res1$weightFisher,method="BH"),digits = 4)
pre_all_res_final1=cbind(all_res1,p.adj1)
all_res_final1 <- pre_all_res_final1[order(pre_all_res_final1$p.adj1),]
write.table(all_res_final1,"summary_topGo_analysis_all_DE_miRs_targets.csv",sep=",",quote=F)
Has anyone had a similar problem with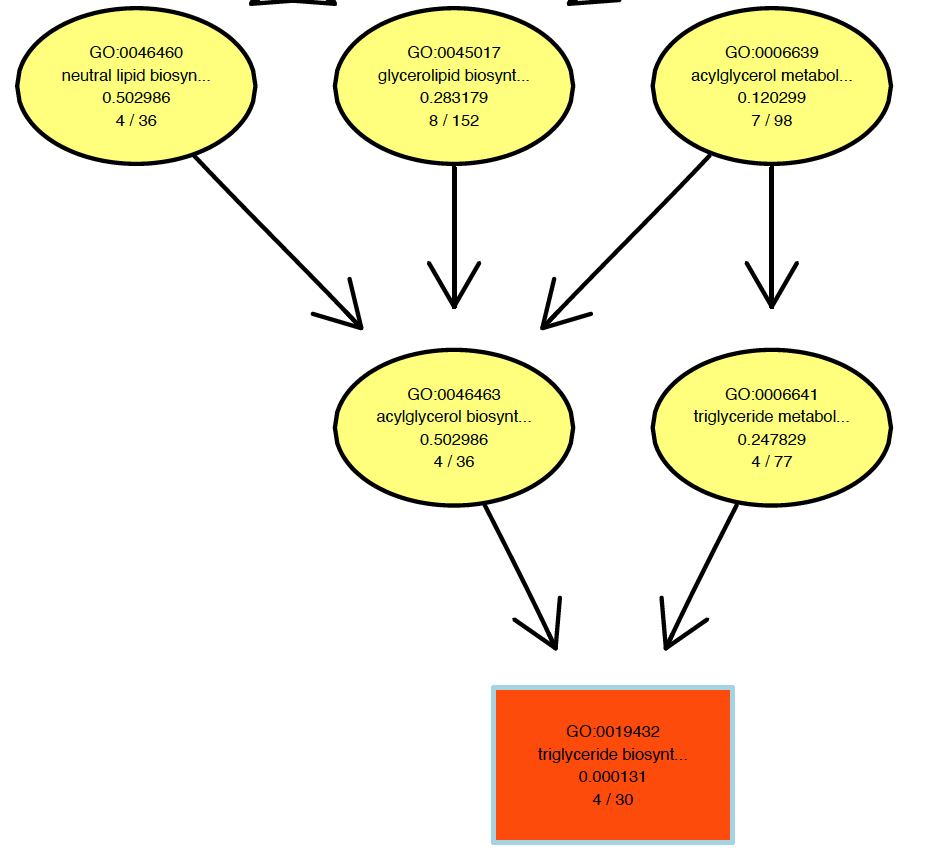
showSigOfNodes? The GO term definitions are getting cut off. I know they are long but I would at least like the option to display the full definition. Here is an example of my problem:Hi, have you found a solution to this?
Did you not notice that this has been answered as part of this thread that you posted in?
Crossposted on biostars: https://www.biostars.org/p/440805/#440807. Could you show
head(all_res1)andall_res1$Nameor similar where the terms are stored?head(all_res1) givesand
all_res1$NameisNULLFor example the
GO:0045944 positive regulation of transcription by ...should be written in the full name likeGO:0045944 positive regulation of transcription by RNA polymerase II