Entering edit mode

I got the network with the WGCNA package and then imported the network into Cytoscape. I applied the analyzer network to obtain the "degree" (I assume that this is the degree of connectivity that will allow me to choose the hub genes.) but the results seem strange to me and I don't know where the error could be, could you please give me some guidance in this regard? Thank you very much in advance
Code should be placed in three backticks as shown below
# R studio V 4.2.3
#loading data
datExpr <- read.csv("TPM_Zscore_Ready.csv", header=TRUE, sep=',')
colnames(datExpr)
# Manipulate file so it matches the format WGCNA needs
row.names(datExpr) = datExpr$Genes
datExpr$Genes = NULL
datExpr = as.data.frame(t(datExpr)) # now samples are rows and genes are columns
dim(datExpr)
# detect outlier genes
gsg = goodSamplesGenes(datExpr, verbose = 3)
gsg$allOK
summary(gsg)
## If the last statement returns TRUE, all genes have passed the cuts. If not, is removed the offending genes and samples from the data.
#datExpr <- datExpr[gsg$goodGenes == TRUE,]
####### Uploading the Trait file ################
#Create an object called "datTraits" that contains your trait data
datTraits = read.csv("Traits_ready.csv")
#form a data frame analogous to expression data that will hold the traits.
rownames(datTraits) = datTraits$Sample
datTraits$Sample = NULL
table(rownames(datTraits)==rownames(datExpr))
# You have finished uploading and formatting expression and trait data
# Expression data is in datExpr, and the corresponding traits are in datTraits
datTraits<-datTraits #[,-1]
##########clustering data and traits
# Calculate sample distance and cluster the samples
# cluster samples
sampleTree2 <- hclust(dist(datExpr), method = "average")
# Convert traits to a color representation: white means low, red means high, grey means missing entry
traitColors <- numbers2colors(datTraits, signed = FALSE)
# Choosing a soft-threshold to fit a scale-free topology to the network
# Call the network topology analysis function
powers = c(c(1:10), seq(from =12, to=50, by=2))
sft = pickSoftThreshold(datExpr,
dataIsExpr = TRUE,
powerVector = powers,
corFnc = cor,
corOptions = list(use = 'p'),
verbose = 5,
networkType = "signed")
sft.data <-sft$fitIndices
write.table(sft, file = "sft_result.txt", sep = ",", quote = FALSE, row.names = T)
# Turn data expression into topological overlap matrix (TOM)
softPower = 40
# Generating adjacency and TOM diss/similarity matrices based on the selected soft-power"
#Calculation of the topological overlap matrix, and the corresponding dissimilarity, from a given adjacency matrix.
adjacency <- adjacency(datExpr, type = "signed", power = softPower)
TOMadj <- TOMsimilarity(adjacency)
dissTOM <- 1- TOMadj
dim(dissTOM)
#save(dissTOM, softPower, file="table_DissTOM.RData")
#load("table_DissTOM.RData")
# Construct modules by Clustering using TOM
# Call the hierarchical clustering function. There is used the pearson correlation (average)
hclustGeneTree <- hclust(as.dist(dissTOM), method = "average")
# Make the modules larger, so set the minimum higher
minModuleSize <- 30
# Module ID using dynamic tree cut
dynamicMods <- cutreeDynamic(dendro = hclustGeneTree,
distM = dissTOM,
deepSplit = 2, pamRespectsDendro = FALSE,
minClusterSize = minModuleSize)
table(dynamicMods)
length(table(dynamicMods))
# Convert numeric labels into colors
dynamicColors = labels2colors(dynamicMods)
#sort(table(dynamicColors), decreasing = TRUE) # get the number of genes by color
table(dynamicColors)
# Merge modules
# Calculate eigengenes
dynamic_MEList <- moduleEigengenes(datExpr, colors = dynamicColors)
dynamic_MEs <- dynamic_MEList$eigengenes
# Calculate dissimilarity of module eigengenes
dynamic_MEDiss <- 1-cor(dynamic_MEs)
# Cluster module eigengenes
dynamic_METree <- hclust(as.dist(dynamic_MEDiss), method = "average")
# Plot the hclust
sizeGrWindow(7,6)
plot(dynamic_METree, main = "Dynamic Clustering of module eigengenes",
xlab = "", sub = "")
# Merge close modules
### height cut of 0.2, corresponding to correlation of 0.80, to merge module with 90% of similarity
dynamic_MEDissThres <- 0.2
# Plot the cut line
abline(h = dynamic_MEDissThres, col = "red")
# Call an automatic merging function
merge_dynamic_MEDs <- mergeCloseModules(datExpr, dynamicColors, cutHeight = dynamic_MEDissThres, verbose = 3)
# The Merged Colors
dynamic_mergedColors <- merge_dynamic_MEDs$colors
# Eigen genes of the new merged modules
mergedMEs <- merge_dynamic_MEDs$newMEs
mergedMEs
write.table(merge_dynamic_MEDs$oldMEs,file="oldMEs.txt")
write.table(merge_dynamic_MEDs$newMEs,file="newMEs.txt")
#===============================================================================
# Export of networks (for me only new MEs) to external software (Cytoscape)
#===============================================================================
for (i in 1:length(merge_dynamic_MEDs$newMEs)){
modules = c(substring(names(merge_dynamic_MEDs$newMEs)[i], 3));
genes = colnames(datExpr)
inModule = is.finite(match(dynamic_mergedColors,modules))
modGenes = genes[inModule]
modTOM=dissTOM[inModule,inModule]
dimnames(modTOM)=list(modGenes,modGenes)
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("output_for_cytoscape/merge_CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),
nodeFile = paste("output_for_cytoscape/merge_CytoscapeInput-nodes-", paste(modules, collapse="-"), ".txt", sep=""),
weighted = TRUE, threshold = -1, nodeNames = modGenes, nodeAttr = dynamicColors[inModule]);
}
#===============================================================================
# Networks to Cytoscape
#===============================================================================
library(RCy3)
cytoscapePing () # make sure cytoscape is open
cytoscapeVersionInfo ()
# cytoscapeVersion "3.10.0"
#### Module white
# use the connection with Cytoscape through the 'RCy3' package
#loading file from cyt
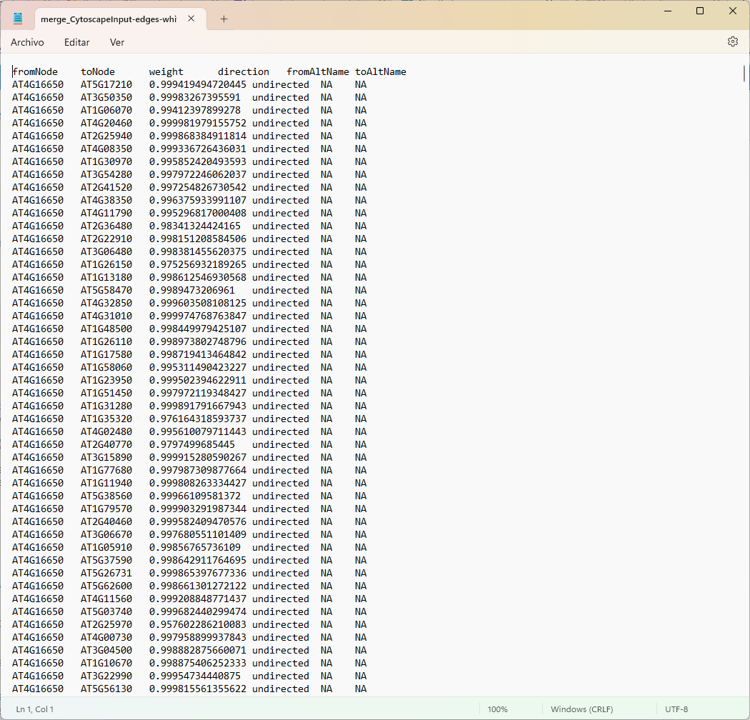
edge <- read.delim("merge_CytoscapeInput-edges-white.txt")
colnames(edge)
# it need to modify colnames
colnames(edge) <- c("source", "target", "weight", "direction","fromAltName", "toAltName")
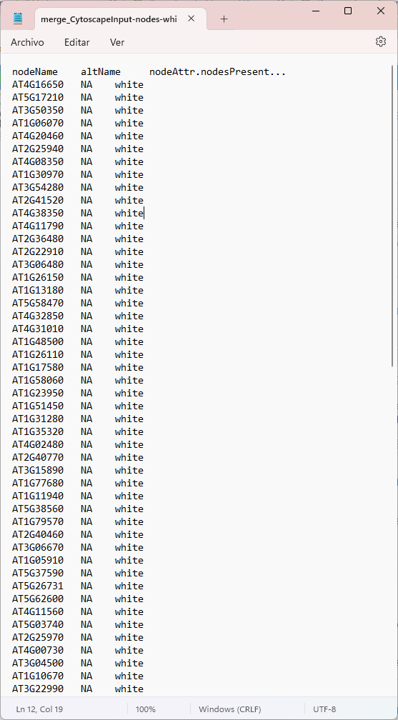
node <- read.delim("merge_CytoscapeInput-nodes-white.txt")
colnames(node)
# it need to modify colnames
colnames(node) <- c("id" , "altName", "node_attributes")
createNetworkFromDataFrames(node,edge, title="white network", collection="DataFrame white")
#createNetworkFromDataFrames(node,edge[1:50,], title="white network", collection="DataFrame white")
#use other pre-set visual style
setVisualStyle('Marquee')
# set up my own style
style.name = "myStyle"
defaults <- list(NODE_SHAPE="ellipse",
NODE_SIZE=30,
EDGE_TRANSPARENCY=120,
NODE_LABEL_POSITION="W,E,c,0.00,0.00")
nodeLabels <- mapVisualProperty('node label','id','p')
nodeFills <- mapVisualProperty('node fill color','node_attributes','d',c("A","B"), c("#FF9900","#66AAAA"))
arrowShapes <- mapVisualProperty('Edge Target Arrow Shape','interaction','d',c("activates","inhibits","interacts"),c("Arrow","T","None"))
edgeWidth <- mapVisualProperty('edge width','weight','p')
createVisualStyle(style.name, defaults, list(nodeLabels,nodeFills,arrowShapes,edgeWidth))
setVisualStyle(style.name)
Edges
nodes
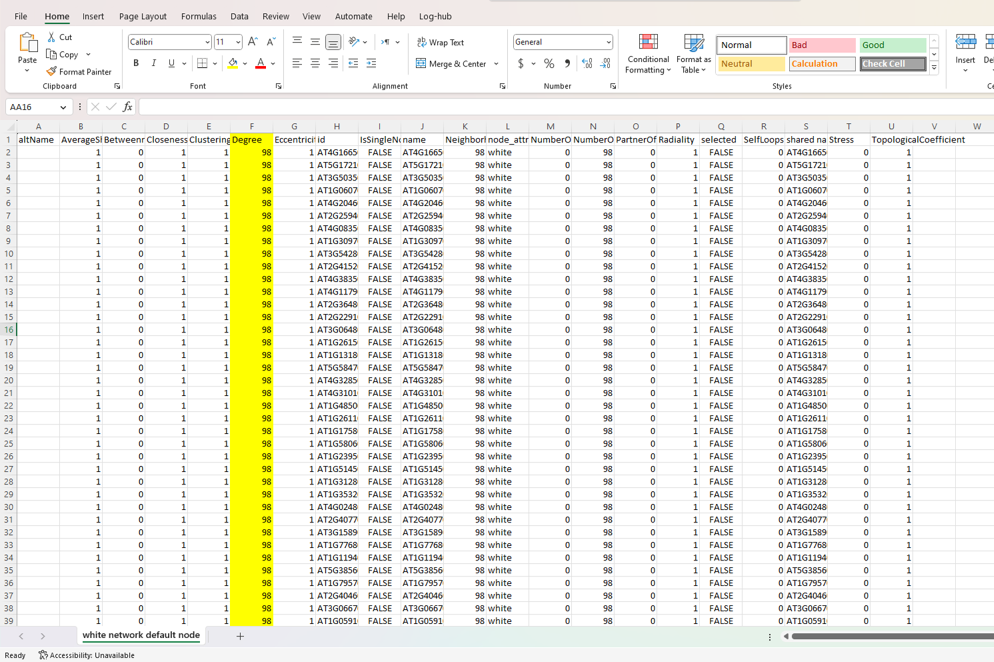
Results from analyzer network

Thank mr If your suspicions are right, how could I apply your respective recommendations?
there are file enter link description here
Yep. The screenshot of the network is pretty clear :)
Everything is connected to everything, so all the degree values will be the same.
You need to delete edges first, then consider degree calculations.
Please, Could you give more detail about the procces?
Thank you very much
It is up to you to choose an edge weight threshold, e.g., >0.999, and then delete the edges below that threshold. Cytoscape and RCy3 do not have an opinion on the threshold you choose. That is a scientific question that only you can answer. You might choose a threshold to give you the top-n edges (where you pick n) or the top n% of edges, or a threshold that results in a degree distribution that is interesting or useful. Totally up to you.
Only after deleting edges will you have degree values that are different across your nodes.
Just to be clear. This is not an RCy3. Each of your nodes has 98 edges connected to it. So, Cytoscape's Network Analyzer function is reporting to you that each node has a degree of 98. That is accurate. You have prune edges (by some method you choose) in order to reduce the degree per node.
ok, thank you very much for your recommendations!