
Hello,
I want to analyze the differentially expressed genes (DEG) of 3 cell populations between tumor and healthy blood.
I have bulk RNA-seq data as a txi matrix containing abundance, counts and length matrixes for 30 samples.
I have a metadata file that indicates for each sample:
- sample ID
- Populations (1,2,3)
- Patient (1,2,3,4,5,6,7,8,9,10)
- Tissu (tumor or healthy blood)
- batch (sequencing n°1 or n°2)
- Tumor stage (2 or 3)
- Tumor type (histological sub-categories of the tumor: 4 values possible including "healthy" for the blood samples)
batch 1: 4 tumors x 3 populations = 12 samples
batch 2 : 1 tumor x 3 populations + 5 blood x 3 populations = 18 populations
The tumor of the batch n°2 seems to clusterize apart from all other samples, so I would like to remove the batch effect due to sequencing runs (see below).
Is it possible even if I don't have blood samples in the first sequencing run?
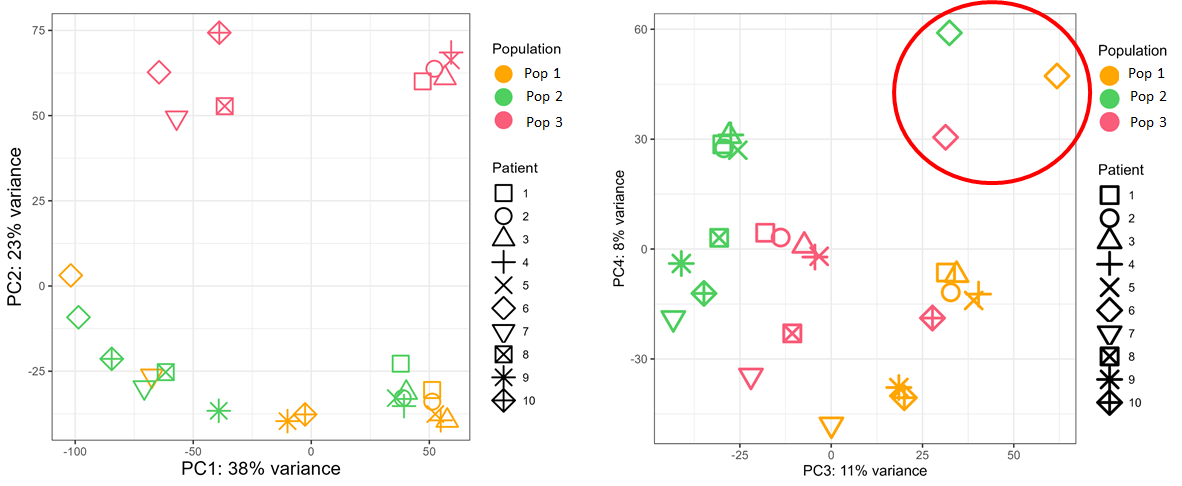
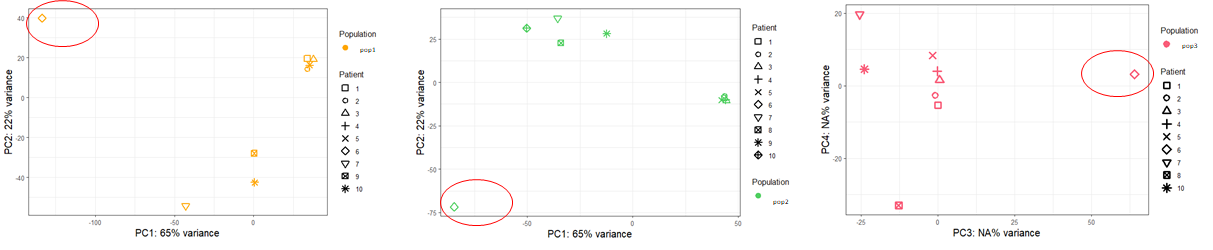
I tried to apply the combat-seq algorithm from sva package (see below) but I face a problem: The code runs well and ends for txi$abundance in <1min, but for the txi$counts, the code runs but never ends, it keeps running even after several hours.
Population <- (factor(meta2$Population))
Tissu <- (factor(meta2$Tissu))
Tumor_type <- (factor(meta2$Tumor_type))
Grade <- (factor(meta2$Grade))
covar_mat <- cbind(Population, Tissu, Tumor_type, Grade)
txi$abundance <- sva::ComBat_seq(txi$abundance, batch=metadata$batch, covar_mod=covar_mat)
Output: Found 2 batches Using null model in ComBat-seq. Adjusting for 4 covariate(s) or covariate level(s) Estimating dispersions Fitting the GLM model Shrinkage off - using GLM estimates for parameters Adjusting the data
txi$counts <- sva::ComBat_seq(txi$counts, batch=metadata$batch, covar_mod=covar_mat)
Output: Found 2 batches Using null model in ComBat-seq. Adjusting for 4 covariate(s) or covariate level(s) Estimating dispersions Fitting the GLM model Shrinkage off - using GLM estimates for parameters Adjusting the data but it keep running and never ends.
I have the same issue even if I include only the 2 factors "Population, Tissu" in the covar_mod.
However, if I put group=Tissu instead of covar_mod, the process ends even for the counts matrix but I completely lose the biological effect due to population identity, so I can't do that.
Would you have an idea why it does not work for the counts matrix?
I understood by reading other posts that combat-seq needs the raw counts, so I need it to work on the counts matrix...
Finally, I tried DESeq2 without combat-seq. In the design whether I put "~ Patient + Population" or "~ batch + Population", I obtain exactly the same PCA_plots, I am quite surprised by that. I would have expected a difference even a slight one...
dds <- DESeqDataSetFromTximport(txi, colData = metadata, design = ~ Patient + Population)
#patient and Population have been transformed as factor before running this.
Moreover, when I subset the dds object to focus for example on the population n°1 to compare between blood and tumor, I have a completely different list of DEGs whether I indicate a design "~ batch + Tissu" or "~ Tissu" alone even if the PCA plots are identical between the 2 designs.
( I can't indicate " ~patient + Tissu" because i obtained the following message: Error in checkFullRank(modelMatrix) : the model matrix is not full rank, so the model cannot be fit as specified. One or more variables or interaction terms in the design formula are linear combinations of the others and must be removed.)
dds_pop1 <- dds[, dds$Population == "pop1"]
dds_pop1@colData@listData$batch <- factor(dds_pop1@colData@listData$batch)
dds_pop1@colData@listData$Tissu <- factor(dds_pop1@colData@listData$Tissu)
dds_pop1 <- DESeqDataSet(dds_pop1, design = ~ batch + Tissu) #I can't put ~ Patient + Tissu because I have the linear combination error
dds_pop1_DEG <- DESeq(dds_pop1)
contrast_tissu <- c("Tissu", "breast", "blood")
res_pop1_unshrunken <- results(dds_pop1_DEG, contrast_tissu, alpha = 0.05)
Do I have to trust the DEG with the design that controls the batch effect ? I can't rely on the PCA distribution because it is the same between the design with or without controling batch
Thank you for any help,
Best regards,
Léo

Can you show
colData(dds)?Aside from that,dds_pop1@colData@listData$batchand direct access to slots like that is pretty clunky. Just dodds$batch.Thanks for your reply. Ok for dds$batch
Please find below the colData(dds).
Concerning the DEG, my question is to know whether I should include batch ("~ batch + Tissu") or not ("~ Tissu") in the design, the PCA plot is the same in both cases.