
Hi,
I am trying to do exploratory analysis of AmpliSeq data using a heatmap and a PCA plot (with DESeq2 and ggplot2). To do this I have been following the DESeq2 vignette. I manage to get both plots, but the sample information data (passage and day in my case) seems to be mixed up and I have not been able to find a reason for this.
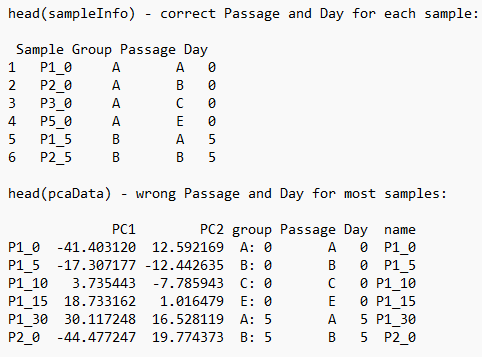
In my data I have a time series with five different days (0, 5, 10, 15, 30), for five different passages of cells (A, B, C, D, E). However, passage D is missing Day 0, and passages D and E are missing day 30.
The sampleInfo for the DESeq looks correct (using head(sampleInfo)), but in the pcaData the sample information is mixed up, that is days and passages have been shifted. When looking at the heatmap, it seems possible that the same mix up has happened to the data.
Please note that I am new to R and appreciate any help I can get.
countdata<-read.csv("All.csv", header=TRUE, row.names=1)
sampleInfo<-read.csv("SI_All.csv", header=TRUE)
matMatrix<-as.matrix(countdata)
rownames <- countdata[,1]
dds <- DESeqDataSetFromMatrix(countData =matMatrix,colData = sampleInfo, design= ~ Group)
dds_norm <- estimateSizeFactors(dds)
normalized_counts <- counts(dds_norm, normalized=TRUE)
write.csv(normalized_counts, file="All_norm_counts.csv")
dds <- DESeq(dds)
res <- results(dds)
DESeq2::plotMA(res,ylim=c(-8,8))
sampleDists <- dist(t(assay(dds)))
library("RColorBrewer")
sampleDistMatrix <- as.matrix(sampleDists)
rownames(sampleDistMatrix) <- paste(dds$Sample, dds$Group, sep="-")
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=colors)
rdds <- rlog(dds)
pcaData <- DESeq2::plotPCA(rdds, intgroup=c("Passage","Day"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2,color=factor(rdds$Passage),shape=factor(rdds$Day))) +
geom_point(size=3) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
coord_fixed()
Session Info:
R version 4.3.1 (2023-06-16 ucrt) Platform: x86_64-w64-mingw32/x64 (64-bit) Running under: Windows 11 x64 (build 22621)
Matrix products: default
locale: 1 LC_COLLATE=English_Sweden.utf8 LC_CTYPE=English_Sweden.utf8 LC_MONETARY=English_Sweden.utf8 [4] LC_NUMERIC=C LC_TIME=English_Sweden.utf8
time zone: Europe/Stockholm tzcode source: internal
attached base packages: 1 stats4 stats graphics grDevices utils datasets methods base
other attached packages:
1 edgeR_3.42.4 limma_3.56.2 DESeq2_1.40.2
[4] SummarizedExperiment_1.30.2 Biobase_2.60.0 MatrixGenerics_1.12.2
[7] matrixStats_1.0.0 GenomicRanges_1.52.0 GenomeInfoDb_1.36.1
[10] IRanges_2.34.1 S4Vectors_0.38.1 BiocGenerics_0.46.0
[13] glmpca_0.2.0 PoiClaClu_1.0.2.1 RColorBrewer_1.1-3
[16] pheatmap_1.0.12 dplyr_1.1.2 ggplot2_3.4.3
loaded via a namespace (and not attached):
1 utf8_1.2.3 generics_0.1.3 bitops_1.0-7 lattice_0.21-8
[5] magrittr_2.0.3 grid_4.3.1 Matrix_1.5-4.1 fansi_1.0.4
[9] scales_1.2.1 codetools_0.2-19 cli_3.6.1 rlang_1.1.1
[13] crayon_1.5.2 XVector_0.40.0 munsell_0.5.0 withr_2.5.1
[17] DelayedArray_0.26.6 S4Arrays_1.0.4 tools_4.3.1 parallel_4.3.1
[21] BiocParallel_1.34.2 colorspace_2.1-0 locfit_1.5-9.8 GenomeInfoDbData_1.2.10
[25] vctrs_0.6.3 R6_2.5.1 lifecycle_1.0.3 zlibbioc_1.46.0
[29] MASS_7.3-60 pkgconfig_2.0.3 pillar_1.9.0 gtable_0.3.4
[33] glue_1.6.2 Rcpp_1.0.11 tibble_3.2.1 tidyselect_1.2.0
[37] rstudioapi_0.15.0 farver_2.1.1 labeling_0.4.3 compiler_4.3.1
[41] RCurl_1.98-1.12
```

Ok, thank you. This is everything from my session:
Check colnames of count data.
I think you are providing these in different order to DESeq2.
You were correct. The order of my samples in my sample info-file did not match the order of the samples in my count data file. Thank you!
Just as a note, if the sample info table had been named (that is
rownames(sampleInfo)notNULL, DESeq2 would have been able to detect the mis-ordering and notify you.