Entering edit mode

Hi all,
Does any one know how to obtain the information under GO class (direct) given a gene list? Ideally I would like to be able to retrieve it with R / API. I have tested several packages but none are able to retrieve this information... Any help would be greatly appreciated!
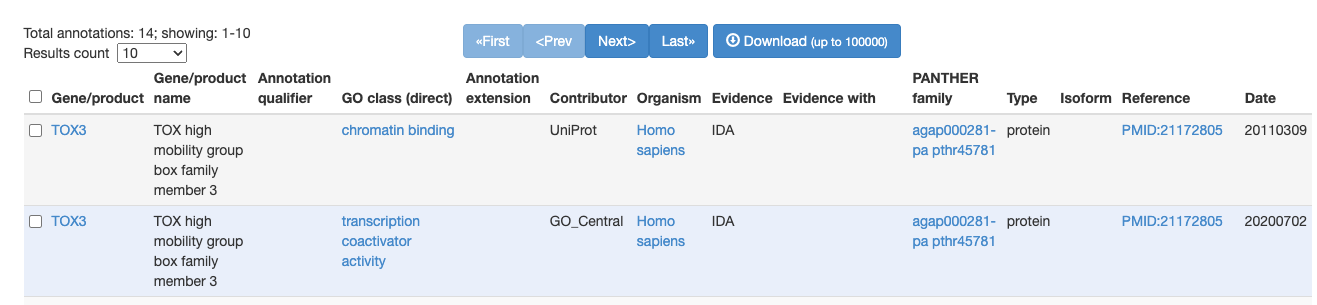
edit: this screenshot is taken from AmiGO Annotations edit2: title updated for clarity

What do you mean by 'the information'? Do you have things in particular that you want?
Sorry I wasn't clear before. What I mean is the all the information associated with the go term, such as description and evidence.
I have since realized that most of this can be done via BiomaRt (under some confusingly named attributes). Included below in case it may help others. e.g.