
Hello,
I have an RNAseq experiment of 10 samples (2 donors, 5 doses). In the PCA plot, we observed that two samples from the same dose group (one from each donor) cluster extremely close together. We are assuming that they were pooled together by mistake. Could there be any other explanation for why these samples are very close to each others?
We suggest that these samples may have been pooled or are technical duplicates, but we want to consider other possibilities.
Questions:
Could there be alternative explanations for why these samples are so similar?
What are the recommended checks to confirm whether they are truly identical or independent?
If confirmed identical, should we exclude them from the analysis, or can we still save them for differential expression since they are from the same dose group and should be replicates of each other?
We have attached the PCA plot for reference. Any guidance would be greatly appreciated.
The PCA plot is attached below.
Thanks in advance.
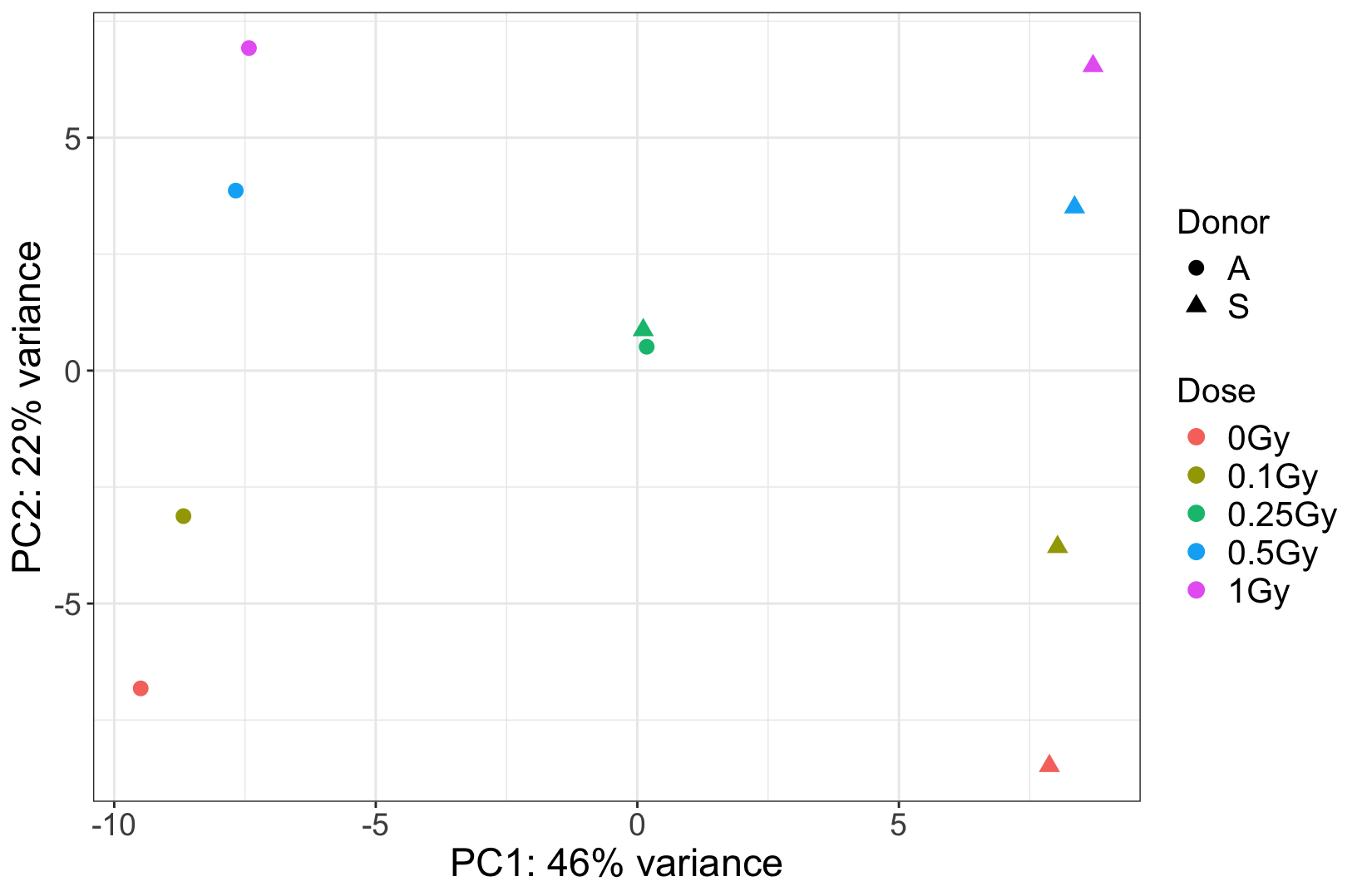

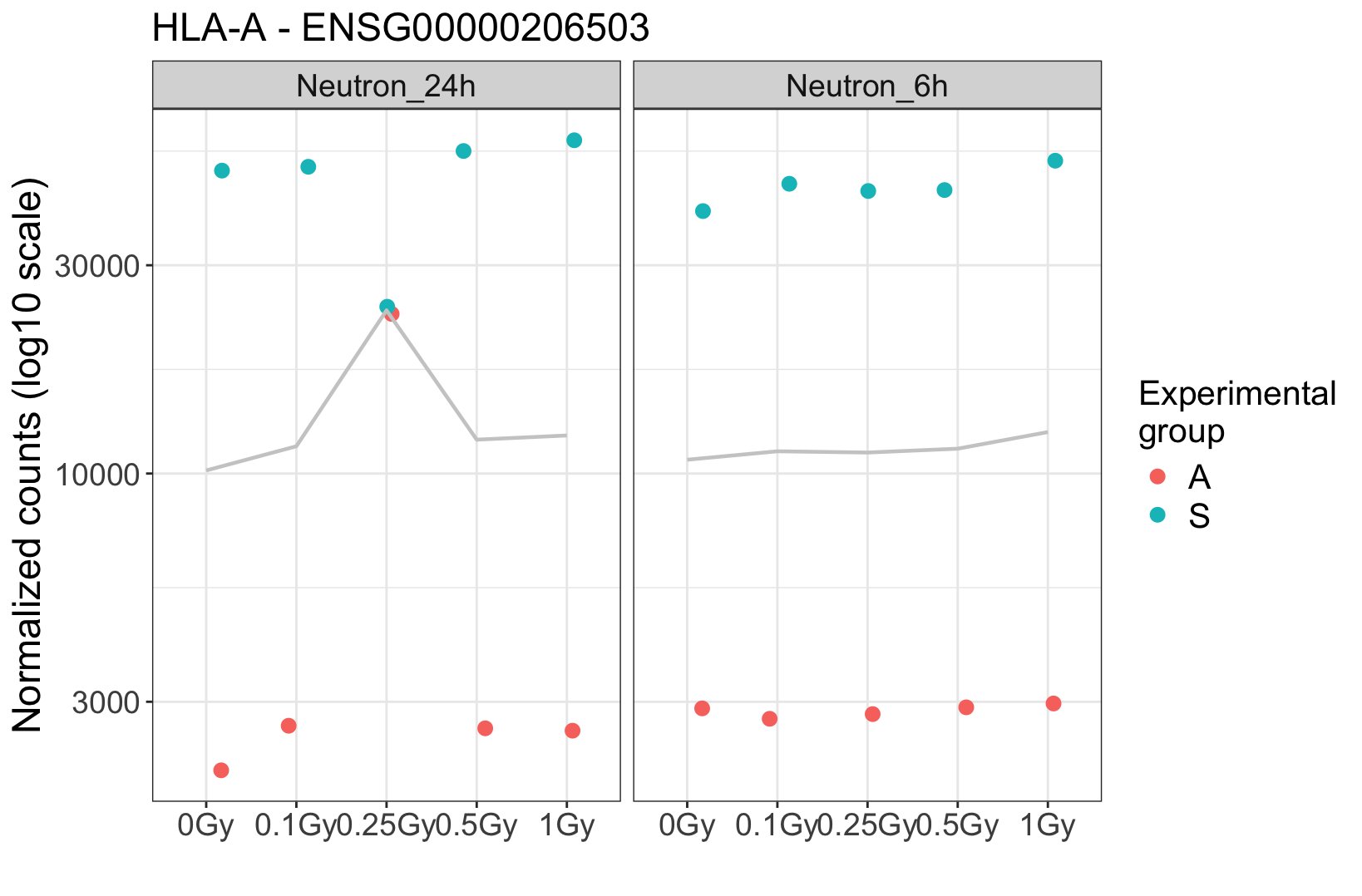
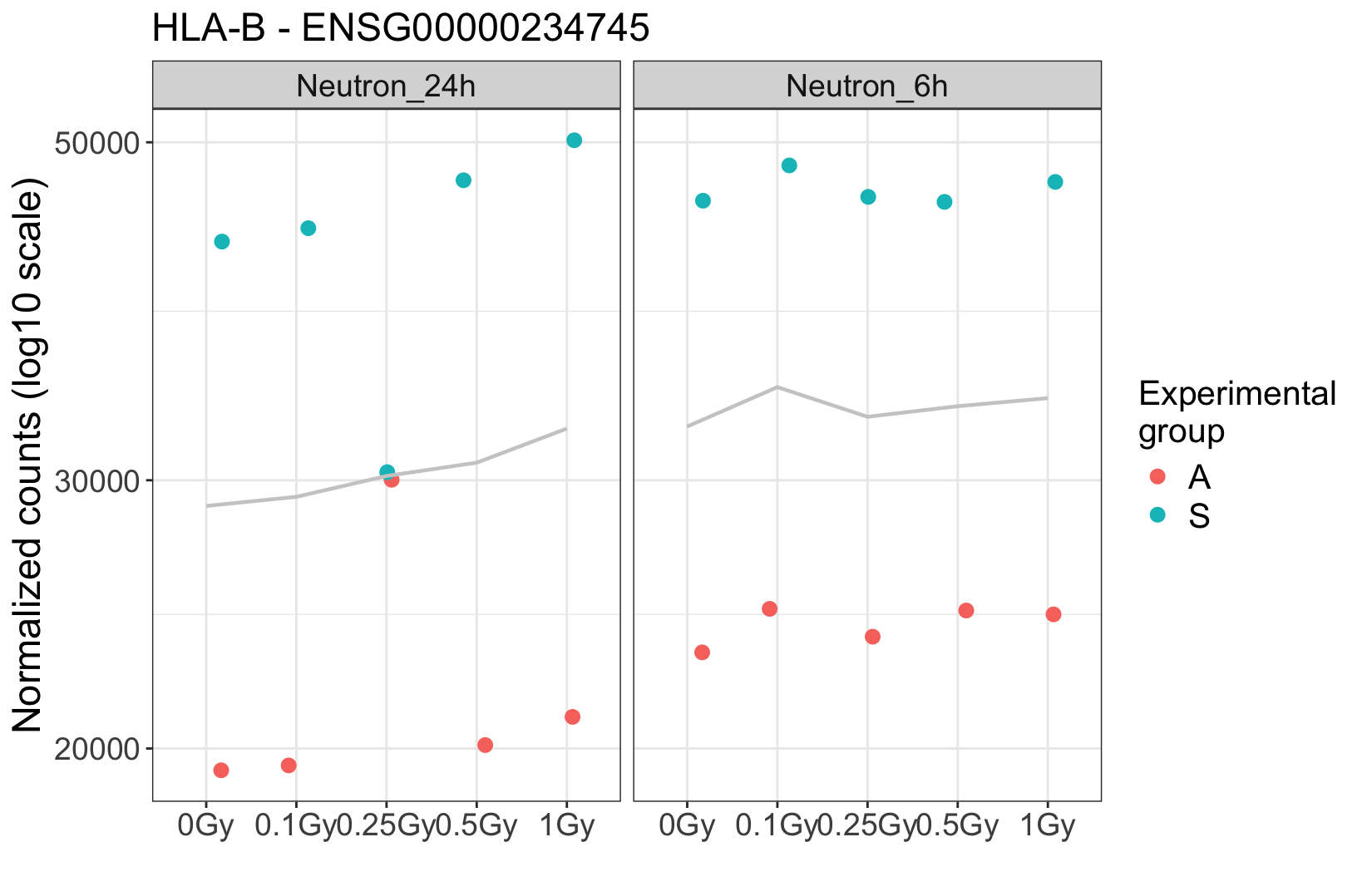 . At this particular dose, it appears that the two suspicious samples were mixed. Additionally, visual inspection in IGV shows many shared SNPs between these two samples that are not shared in the clean ones.
. At this particular dose, it appears that the two suspicious samples were mixed. Additionally, visual inspection in IGV shows many shared SNPs between these two samples that are not shared in the clean ones.
The donor effect is crystal clear, and absent in these suspicious samples. It strongly argues for a technical problem. If you really need some hard facts, maybe find some genes that are known to have lots of person-specific polymorphisms, and then see whether a) in the clear samples these are different between the two donors, and then b) in the suspicious ones you find heterozygosity. Question is if you need this since the overall picture seems so clear.