Entering edit mode

I am getting following error related to read.metharray, while running my Script,
Error in read.metharray(basenames = files, extended = extended, verbose = verbose, :
!anyDuplicated(basenames) is not TRUE
FYI, I made my sample sheet on my own by removing the information of some of the samples for which the data was missing. I have double checked that now my meth_idat folder has exactly the same number of samples each with 2 idat files as mentioned in my sample sheet. But still getting this error.
Any help will be highly appreciated.
Thanks

Hi there, I'm completely new to methylation analysis so apologies for my naivety! I have used the EPIC beadchip (8 sample) for my experiment. I'm also having this issue when I try to read the data into R. The idat files are named like this:
205128000104_R01C01_Grn.idat
205128000104_R01C01_Red.idat
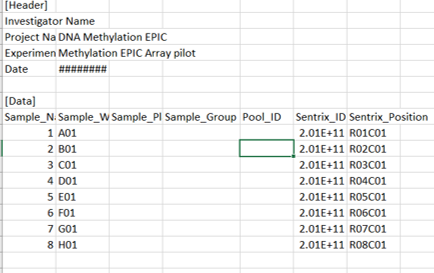
From the above post do you mean I need manually edit the idat file names for it to be recognised? Because doing so would overwrite one of the files. Or have I misunderstood? I have attached a picture of my sample sheet.
Any help would be greatly appreciated.