
I would like to test the zinbwave package with my data, but not sure, what is the correct procedure for that.
We have several experimental conditions/timepoints sequenced by 10x and would like to compare them against each other. They are all in duplicates (two biological replicates for each TP).
How would one go ahead with duplicated? Do I need to merge the two biological replicates into one sample and then continue to work with it as one, or is there a way to classify them as belonging to the same condition, when doing the DE analysis?
Would it be better first to use Seurat to integrate each two conditions (so four samples) to prevent possible batch effects into one integratedData object, keeping the project.Ident in place so they can be compared?
thanks

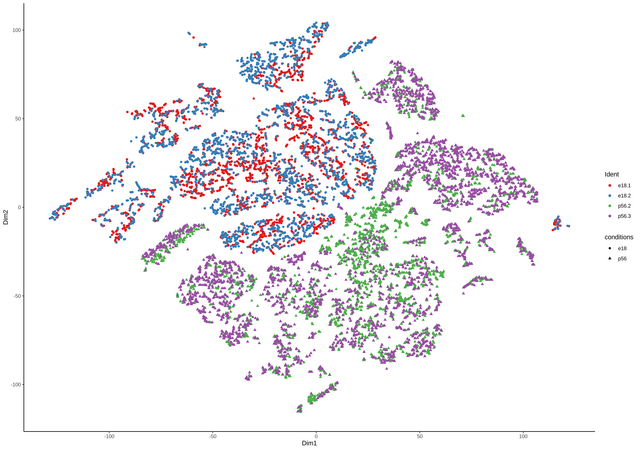
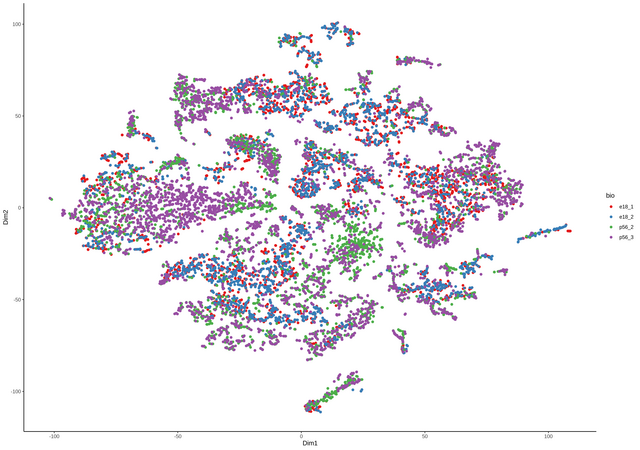
continuing the discussion from github, thanks Davide for the great package. comment from Davide
So I was wondering how to proceed with my data-
I have had already the data merged in
Seuratand than converted to asingleCellExperimentobject containing all four samples. TheColdatacontains the data from theSeuratobject as well, including the origin (orig.ident) of the samples (see below).I can add another column with the name for the two groups, something like e.g.
the number are the lengths of the four single samples for each group.
Then when running the
zinbwavecommandThis is what I did
Or should I use
K=2as I have two groups?But based on your recommendation, should I try now
Would this be the correct appraoch to compensate for possible bias due to the differences in the samples?
thanks again Assa