Entering edit mode

MOHAMMAD
•
0
@MOHAMMAD-24781
Last seen 3.4 years ago
I am a beginner in R I did Differential expressed gene analysis and I have two questions:
1- depending on the graphs is the differentially expressed genes list reliable or not For further analysis?
I am disappointed because I didn't get the segregation of the samples in the heatmap and PCA1 and PCA2 are low
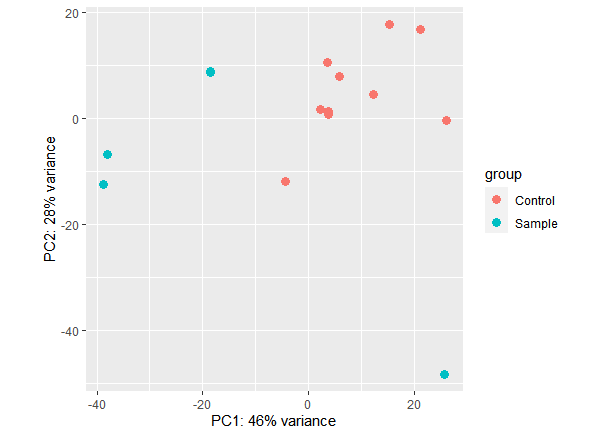
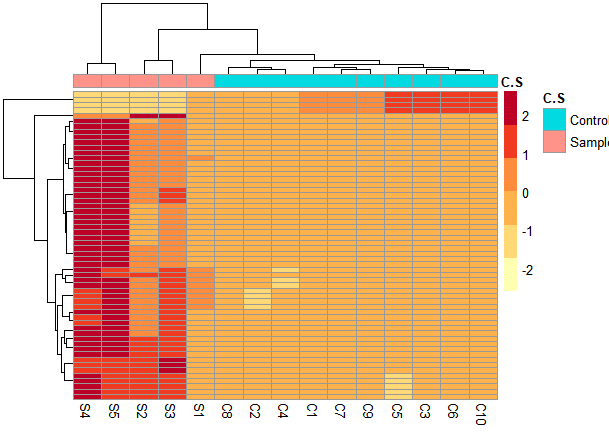
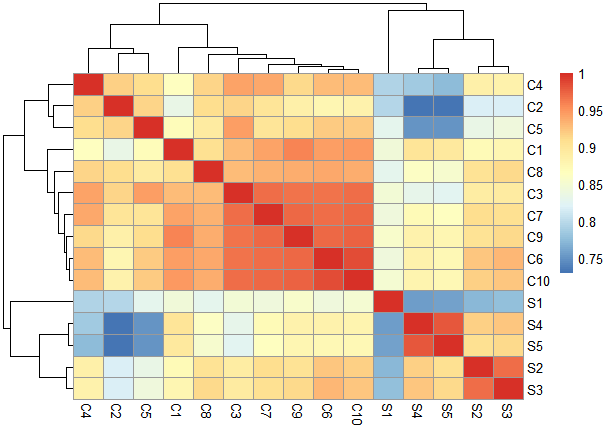

I noticed that the sample S1 varies from other samples resulting in low PCA1 but also different from the 10 controls. is there any way to handle it?!
2- when I am exporting the list of DEGs I get (some genes appears many times) For example, if the gene id is df3t_00100 I got records as following:
df3t_00100
df3t_00100.1
df3t_00100.2
df3t_00100.3
what are those and how can I handle them?
Thank you in advance!

PCA code:
vsd <- vst(dds, blind = T) # Varaiance Stabilizing transformation
plotPCA(vsd, intgroup = "C.S")
2- the organism is plasmodium falicparium
design(dds) <- ~ C.S
dds <- DESeq(dds)
res <-results(dds)
summary(res)
resSort <- res[order(res$padj),]
library("org.Pf.plasmo.db")
geneinfo <- select(org.Pf.plasmo.db, keys=rownames(resSort)[1:20], columns=c("SYMBOL","GENENAME","GO"), keytype="SYMBOL")
geneinfo
gene info returns some repeated genes and some with decimal:
Thanks, you are evidently not following the typical DESeq2 analysis pipeline - you are missing the
lfcShrink()stage. Please take a look at the Quick start.Are you showing all of the output of
geneinfo? There seems to be at least 2 columns missing.Thank you,
can you kindly where should I use ilfcShrink() stage
the geneinfo output is ok its just cut to show gene_id
Hi, regarding lfcShrink, the information is in the Quick start (please see my other comment)