Entering edit mode

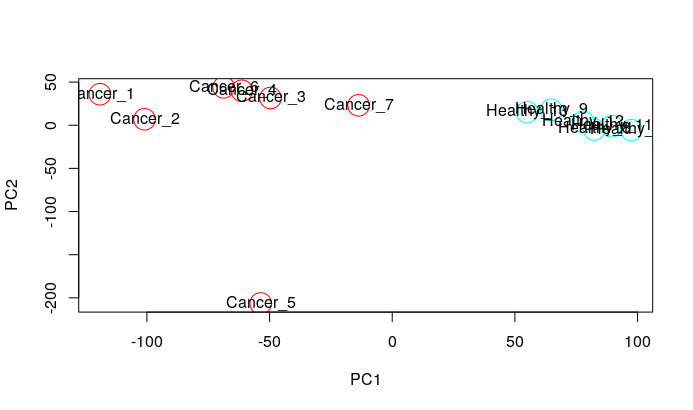
I have done the quality controls like PCA and hierarchical clustering and found an outlier, which is Cancer5.CEL. How can I remove this outlier for Differential Gene Expression Analysis? I don't know the code, please help. Thank you!
my code:
crc.fac<- factor(c(rep("Cancer", 7),rep("Healthy",6)))
crc.df <- data.frame(crc = crc.fac,row.names = paste(crc.fac, rep(1:13, 1), sep = ''))
crc.mData <- data.frame(labelDescription = c("gene regulation"))
crc.mData
crc.pData <- new("AnnotatedDataFrame", data = crc.df, varMetadata = crc.mData)
validObject(crc.pData)
[1] TRUE
list.files(path = ".", pattern = ".CEL")
crc.df <- data.frame(crc.fac, filename = list.files(path =".",pattern=".CEL"),row.names =paste(crc.fac, rep(1:13, 1), sep = ''))
crc.affy <- read.affybatch(filename = list.files(path =".",pattern=".CEL", full.names = TRUE),
+ phenoData = crc.pData)
View(crc.affy)
crc_calls.eSet <- mas5calls.AffyBatch(crc.affy)
crc_calls.mx <- exprs(crc_calls.eSet)
crc.eSet <- rma(crc.affy)
crc_log2.mx <- exprs(crc.eSet)
head(crc_log2.mx)
boxplot(as.data.frame(crc_log2.mx), xlab = "", ylab = "Log2 rma signal", las = 2, main = "Sample Distributions")
crc_P_rate.nv <- apply(crc_calls.mx == "P", 2, sum) / nrow (crc_calls.mx)
quality controls:
check potential physical defects in the arrays
image(crc.affy[, 1])
PCA
pca <- prcomp(t(crc_log2.mx))
eigs <- pca$sdev^2
varexplained <- eigs/sum(eigs)
varexplained
barplot(varexplained * 100, ylab="% variance explained", xlab="principal components")
box()
plot(pca$x[, 1], pca$x[, 2], col=rep(rainbow(2), each=7,6), xlab="PC1", ylab="PC2", cex=3)
text(pca$x[, 1], pca$x[, 2], labels = colnames(crc_log2.mx))
I have found the below remove outlier code, but it seem didn't work on my case, please help!!
remove_outliers <- function(x, na.rm = TRUE, ...) {
qnt <- quantile(x, probs=c(.25, .75), na.rm = na.rm, ...)
H <- 1.5 * IQR(x, na.rm = na.rm)
y <- x
y[x < (qnt[1] - H)] <- NA
y[x > (qnt[2] + H)] <- NA
y
}
# Removes all outliers from a data set
remove_all_outliers <- function(df){
# We only want the numeric columns
a<-df[,sapply(df, is.numeric)]
b<-df[,!sapply(df, is.numeric)]
a<-lapply(a,function(x) remove_outliers(x))
d<-merge(a,b)
d
}
# Removes all outliers from a data set
remove_all_outliers1 <- function(df){
# We only want the numeric columns
df[,sapply(df, is.numeric)] <- lapply(df[,sapply(df, is.numeric)], remove_outliers)
df
}
remove_all_outliers2 <- function(df){
df[] <- lapply(df, function(x) if (is.numeric(x))
remove_outliers(x) else x)
df
}
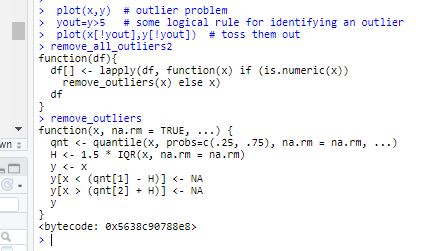
If I can't exclude outlier, it may affect the further processing "Differential gene expression analysis" Please help!

Actually, I didn't know what remove outlier code can be applied to my case.