
My RNA-seq data has a large batch effect, known (the sequencing depth is different for the first two samples from the rest) and unknown.
first, I plotted a histogram on the count data.
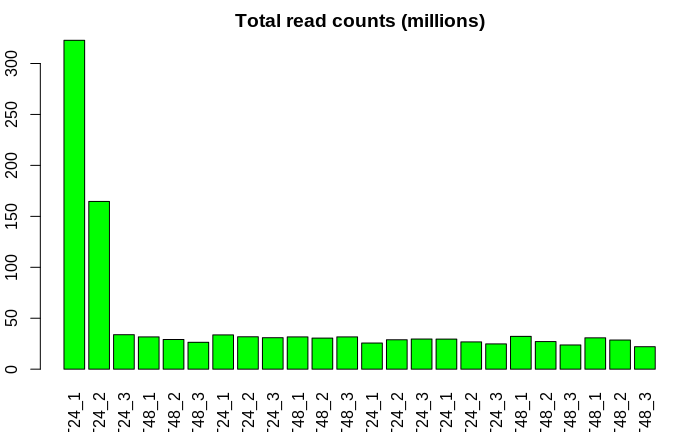
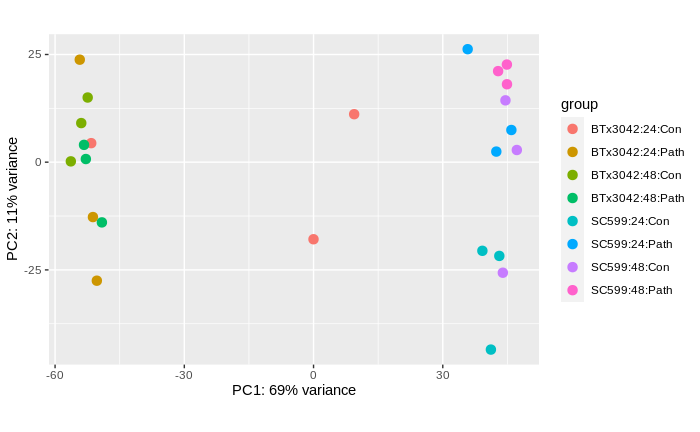
then I did deseq using this code and checked the pca plot it is mixed up and the two samples with large counts are plotted between the two genotypes.
dds=DESeqDataSetFromMatrix(countData = countData, colData = colData,
design = ~Genotype+Rep+treatment+Time+Genotype*treatment+Genotype*Time+treatment*Time)
dds <- estimateSizeFactors(dds)
dds_5=dds[rowMeans(counts(dds,normalized=TRUE))>=5,]
dds_5 <- estimateDispersions(dds_5)
dds = nbinomLRT(dds_5, maxit=100,
full = formula(~Genotype+Rep+treatment+Time+Genotype*treatment+Genotype*Time+treatment*Time),
reduced = formula(~ Genotype+Rep+treatment+Time))
sizeFactors(dds)
sizeFactor between the first two samples, the last sample, and the others have huge variation.

rld = rlog(dds, blind = FALSE)
plotPCA(rld, intgroup= c("Genotype","Time", "treatment"))
They are mixed up even the same rep never get together
Then I tried svseq using the code below. I was switching the filter of rowMeans from 5 through 500 and the maxit from 100 through 100, 000, I am getting "___ rows did not converge in beta, labeled in mcols(object)$fullBetaConv. Use larger maxit argument with nbinomLRT" all the time. but I created batch as a factor based on what I see in the data. I tried with and without it.
Batch = c("1", "2", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3", "3")
dds=DESeqDataSetFromMatrix(countData = countData, colData = colData,
design = ~Batch+Genotype+Rep+treatment+Time+Genotype*treatment+Genotype*Time+treatment*Time)
dds$treatment <- factor(dds$treatment, levels = c("Con","Path"))
dds$Genotype <- factor(dds$Genotype, levels = c("BTx3042","SC599"))
dds$Time <- factor(dds$Time, levels = c("24","48"))
dds$Batch <- factor(dds$Batch, levels = c("1", "2", "3"))
mod = model.matrix(~ Batch+Genotype+Rep+treatment+Time+Genotype*treatment+Genotype*Time+treatment*Time, data=colData(dds))
mod0 = model.matrix(~1,data=colData(dds))
dds = estimateSizeFactors(dds)
countDat = counts(dds,normalized=TRUE)
countDat=countDat[rowMeans(countDat)>50,]
svseq = svaseq(countDat,mod,mod0, n.sv=3)
ddssva = dds
ddssva$SV1 <- svseq$sv[,1]
ddssva$SV2 <- svseq$sv[,2]
ddssva$SV3 <- svseq$sv[,3]
design(ddssva) = ~ SV1 + SV2 +SV3+
Batch+ Rep+Genotype+treatment+Time+Genotype*treatment+Genotype*Time
ddssva <- estimateSizeFactors(ddssva)
ddssva <- estimateDispersions(ddssva)
ddssva = nbinomLRT(ddssva, maxit=1000,
reduced = formula(~ Batch+Genotype+Rep+treatment+Time))
Michael Love Kevin Blighe Please help me.