
Hello everybody,
I created a simple and a complex heatmap comparing gene expressions in two groups (control vs SAH) and noticed that the z-score color coding of the complex heatmap does not seem to fit the z-scores.
I picked one special molecule in the heatmap to display the differences in the simple and the complex heatmaps. Here you can find the log fold change values:

And the corresponding z-scores:
lcpm.df.scaled <- t(apply(lcpm.df, 1, scale))

Simple heatmap:
library(gplots)
Control.vs.SAH.topgenes <- Control.vs.SAH$ENTREZID[1:100]
i <- which(v$genes$ENTREZID %in% Control.vs.SAH.topgenes)
mycol <- colorpanel(1000,"blue","white","red")
heatmap.2(lcpm[i,], scale="row",
labRow=v$genes$SYMBOL[i], labCol=group,
col=mycol, trace="none", density.info="none",
margin=c(8,6), lhei=c(2,10), dendrogram="column")
sessionInfo(R version 4.2.1 (2022-06-23))
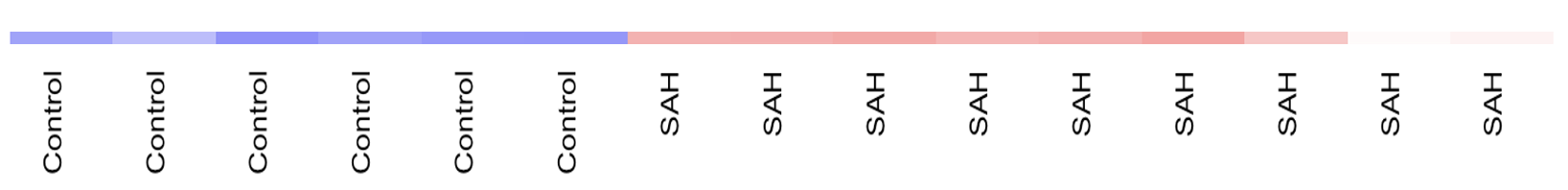
This color-coding makes sense for me.
The problem: It seems as if something went wrong with the z-score color coding in the complex heatmap.
Complex Heatmap:
ha <- HeatmapAnnotation(summary = anno_summary(gp = gpar(fill=2),
height = unit(2, "cm")))
h1 <- Heatmap(lcpm.df.scaled[rows_keep,], cluster_rows = F,
column_labels = colnames(lcpm.df.scaled), name = "Z-Score",
cluster_columns = T)
h2 <- Heatmap(l2_val, row_labels = df.top$SYMBOL[rows_keep],
cluster_rows = F, name = "logFC", top_annotation = ha, col = col_logFC,
cell_fun = function (j, i, x, y, w, h, col)
grid.text(round(l2_val[i, j], 2), x, y))
h3 <- Heatmap(mean, row_labels = df.top$SYMBOL[rows_keep],
cluster_rows = F, name = "AveExpr", col=col_AveExpr,
cell_fun = function (j, i, x, y, w, h, col)
grid.text(round(mean[i,j],2), x, y))
h <- h1+h2+h3
h
On the left you can see the z-score color coding, followed by the log-FC(6.98) and on the right you can see the AveExpr (12.55).
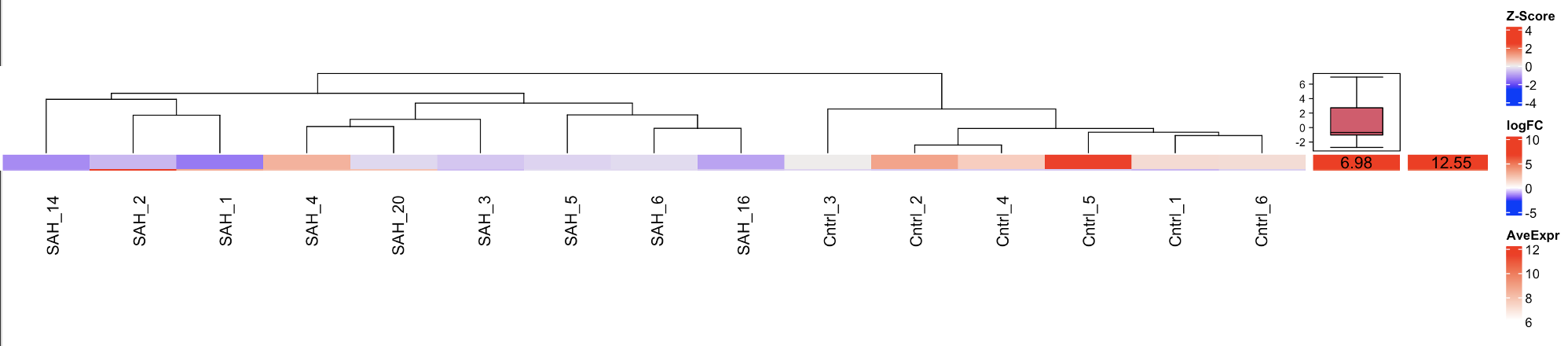
Do you have any idea why the complex heatmap does not really fit the z-scores?
Thank you a lot for your help.

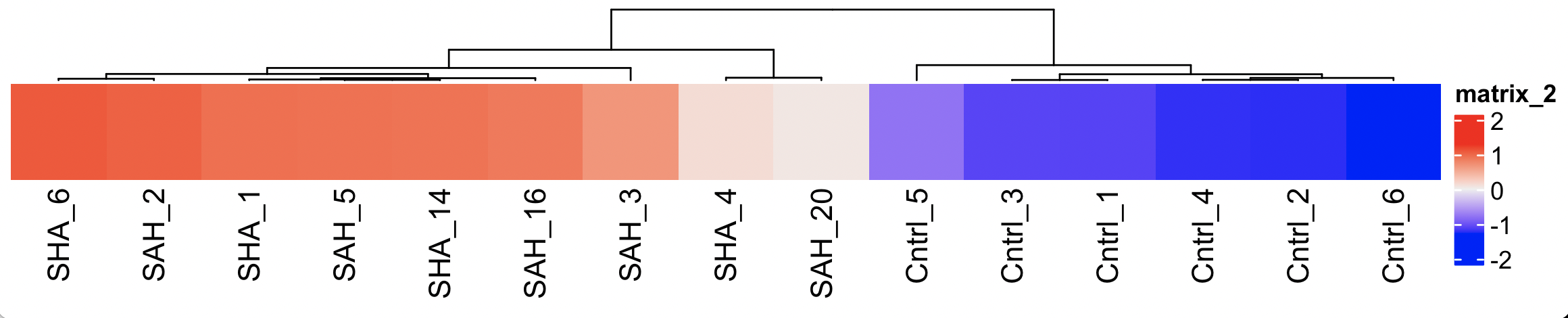

Thank you so much! I added the first 25 genes in a decreasing logFC ranking as h2 on the right side and the corresponding average expressions as h3. It turned out that the z-scores of the actual heatmap h1 were not ranked so that the wrong molecules were matched.