Entering edit mode

Hi,
I want to clarify what is going on under the hood when a user runs DESeq2::estimateSizeFactors() with the controlGenes argument. So, if my understanding is correct, all the steps of size-factor estimation take place, except they are applied only to the genes assigned to controlGenes (except for the final step, which is to apply the calculated size factor to all sample-wise genes) rather than the default of all genes supplied to DESeq2::estimateSizeFactors()—is that right?
A related follow up question: Are there any circumstances in which a user that has spike-in controlGenes for their samples would not want to use them?
Thanks,
Kris

That having said, I recommend to inspect the MA-plots (
plotMA()), at best the ones produced right fromres <- results(dds); plotMA(res)and see how it looks. The plots usually have an arrowhead-like shape and the "tip" of the arrowhead, so the very righthand part of the plot should be nicely centered at y=0. If that is not the case you would a subset of genes for normalization instead of all. Here is an example of a good and a bad plot (made-up data)The good plot:
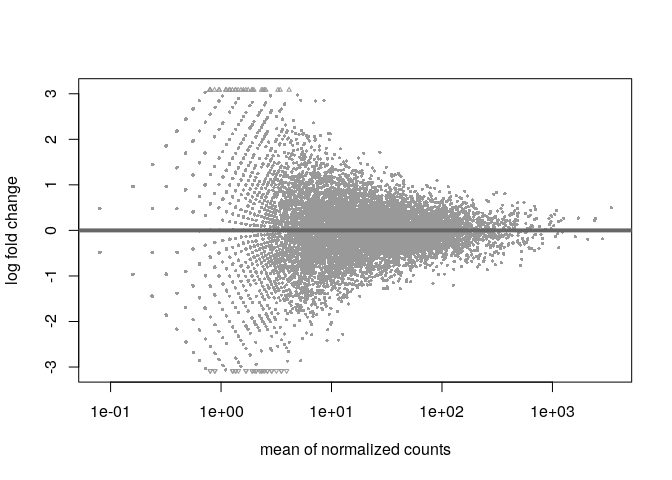
The bad plot:
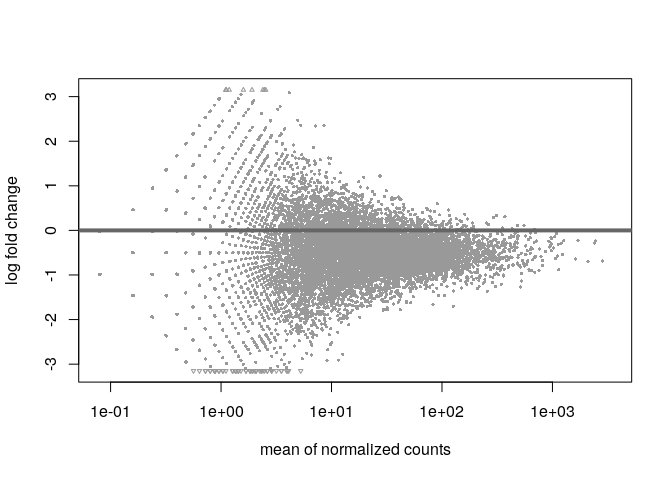
If you get a plot like the second one you can try to use different sets of genes to improve the normalization. If the spikes do not work, then try those genes with overall large baseMeans (like top 10% of genes based on baseMean) to focus on those that are likely stable and not DE.