Entering edit mode

I am trying to align RNA sequencing data using the following code. However, it shows an error killed at the end. It does not specify the reason as well. I initially assumed memory could have been an issue, but the error seems to continue even when the memory issue was sorted in a new system.
This is my code. Any help would be greatly appreciated.
myExons <- exons(TxDb.Hsapiens.UCSC.hg19.knownGene, columns = c("tx_id", "gene_id"))
myExons <- myExons[lengths(myExons$gene_id) == 1]
myExons
dfExons <- as.data.frame(myExons)
SAF <- data.frame(GeneID = dfExons$gene_id, Chr = dfExons$seqnames, Start = dfExons$start,
End = dfExons$end, Strand = dfExons$strand)
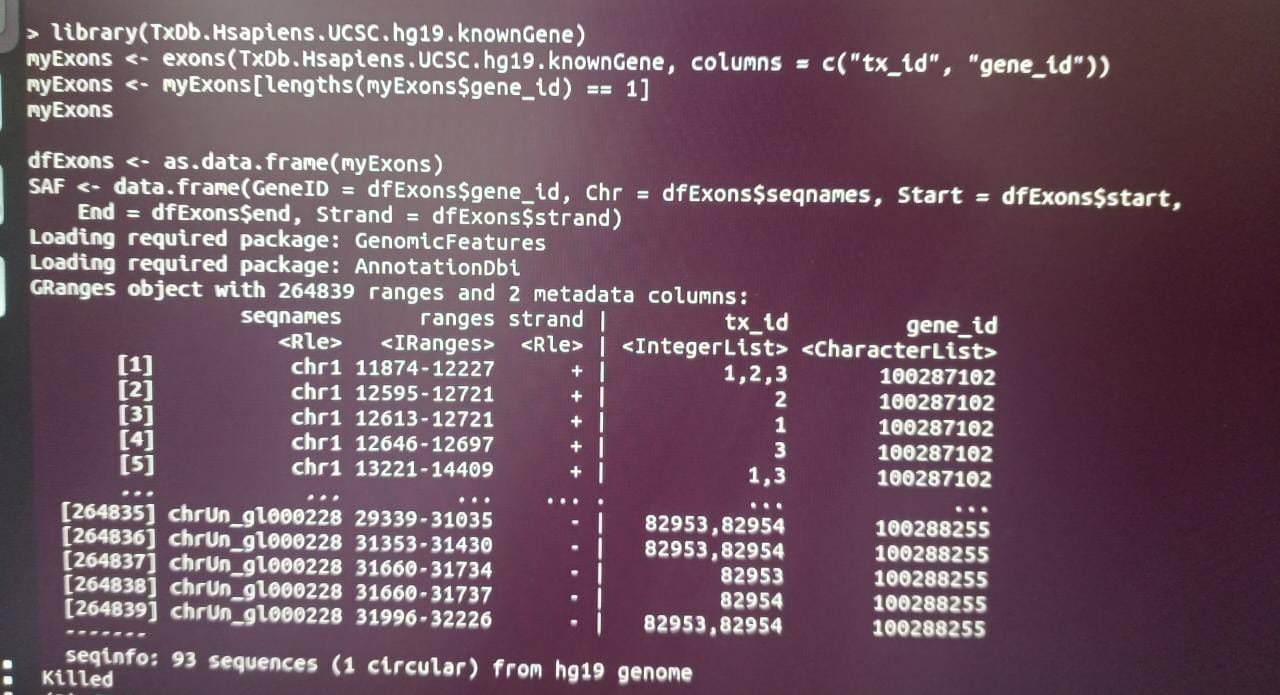

As you can see in the output, some columns in this GRanges object are character vectors, others are lists, so this cannot readily be parsed into a meaningful data.frame, that is why it takes long and crashes. Your code does not do any alignment, so why don't you state what the end goal is so one can suggest better code to avoid these issues. Do you want to make a tx2gene map for tximport?