Entering edit mode

heiko_kin
▴
60
@heiko_kin-23266
Last seen 5.0 years ago
Dear all,
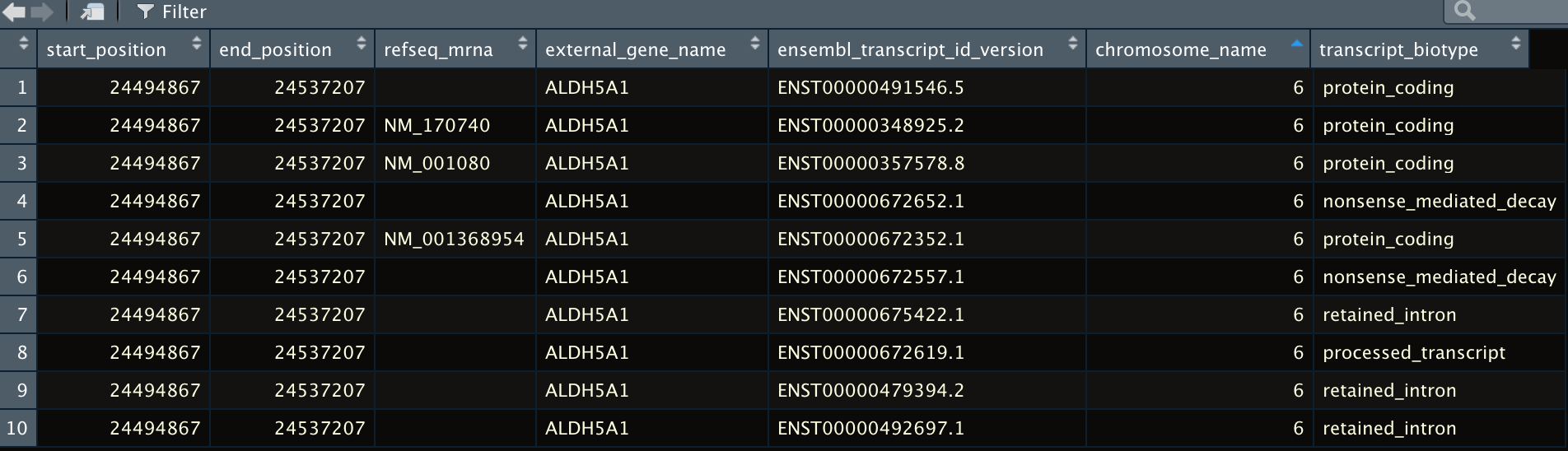
I am trying to extract the canonical coding sequence for a given gene. When using getBM I get a list of several sequences. Is there a way to filter them so I only get the canonical sequence? Thank you all for your help!
library(biomaRt)
mart <- useMart('ensembl',
dataset = 'hsapiens_gene_ensembl',
host = 'useast.ensembl.org')
geneSet <- "ALDH5A1"
result_gene_name <- biomaRt::getBM(attributes = c("start_position","end_position","refseq_mrna","external_gene_name","ensembl_transcript_id_version","transcript_biotype","chromosome_name"),
filters = "external_gene_name",
values = geneSet,
mart = mart)
View(result_gene_name)
sessionInfo()
R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] de_DE.UTF-8/de_DE.UTF-8/de_DE.UTF-8/C/de_DE.UTF-8/de_DE.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] viridis_0.5.1 viridisLite_0.3.0 ggpubr_0.4.0 forcats_0.5.0 stringr_1.4.0 purrr_0.3.4
[7] tidyr_1.1.2 tibble_3.0.4 tidyverse_1.3.0 readr_1.4.0 jsonlite_1.7.2 httr_1.4.2
[13] ggnewscale_0.4.4 drawProteins_1.10.0 cowplot_1.1.0 ggsci_2.9 scales_1.1.1 ggplot2_3.3.2
[19] dplyr_1.0.2 reshape_0.8.8 biomaRt_2.46.0
loaded via a namespace (and not attached):
[1] nlme_3.1-151 fs_1.5.0 lubridate_1.7.9.2 bit64_4.0.5 progress_1.2.2 tools_4.0.3
[7] backports_1.2.1 utf8_1.1.4 R6_2.5.0 mgcv_1.8-33 DBI_1.1.0 BiocGenerics_0.36.0
[13] colorspace_2.0-0 withr_2.3.0 gridExtra_2.3 tidyselect_1.1.0 prettyunits_1.1.1 bit_4.0.4
[19] curl_4.3 compiler_4.0.3 cli_2.2.0 rvest_0.3.6 Biobase_2.50.0 xml2_1.3.2
[25] labeling_0.4.2 askpass_1.1 rappdirs_0.3.1 digest_0.6.27 foreign_0.8-81 rio_0.5.16
[31] pkgconfig_2.0.3 dbplyr_2.0.0 rlang_0.4.9 readxl_1.3.1 rstudioapi_0.13 RSQLite_2.2.1
[37] farver_2.0.3 generics_0.1.0 zip_2.1.1 car_3.0-10 magrittr_2.0.1 Matrix_1.3-0
[43] Rcpp_1.0.5 munsell_0.5.0 S4Vectors_0.28.1 fansi_0.4.1 abind_1.4-5 lifecycle_0.2.0
[49] stringi_1.5.3 carData_3.0-4 plyr_1.8.6 BiocFileCache_1.14.0 grid_4.0.3 blob_1.2.1
[55] parallel_4.0.3 crayon_1.3.4 lattice_0.20-41 splines_4.0.3 haven_2.3.1 hms_0.5.3
[61] pillar_1.4.7 ggsignif_0.6.0 stats4_4.0.3 reprex_0.3.0 XML_3.99-0.5 glue_1.4.2
[67] data.table_1.13.4 modelr_0.1.8 vctrs_0.3.6 cellranger_1.1.0 gtable_0.3.0 openssl_1.4.3
[73] assertthat_0.2.1 openxlsx_4.2.3 broom_0.7.3 rstatix_0.6.0 AnnotationDbi_1.52.0 memoise_1.1.0
[79] IRanges_2.24.1 ellipsis_0.3.1

You could extract the so-called APPRIS score via biomaRt. From the abstract of the APPRIS paper:
As always, there will be ambiguous calls though, so not every gene has a single principal isoform, some have multiple, and probably some have none.
Thank you for pointing that out! I will use Kevin's approach and go for the longest CDS sequence.
Fair enough, I do not think though that there is any evidence that the length alone has any meaning. If it was then efforts such as APPRIS would not have collected extensive experimental data rather than just sorting transcripts by length.
I gave it some more thought and came up with another idea - I can retrieve the "transcript_mane_select" information from the getBM function... as far as I was able to check, it actually suits with the "canonical" transcripts that I was looking for. Anyways, for my application it should be alright. Thanks again and happy new year to you!