Entering edit mode

Hi, Dr love.
I post a question about weird MAplot or volcano plot of DESeq2 diff result and also in biostar. ATpoint give a useful answer about too many 0 count genes and prefiltering.
It seems that too many 0 count genes makes lfc shrink have a probelm. And I find the apeglm and ashr result is very different. I am wondering whether you can give me some advice or which algorithms I shoud choose in this condition.
I have prefilter 0 count gene
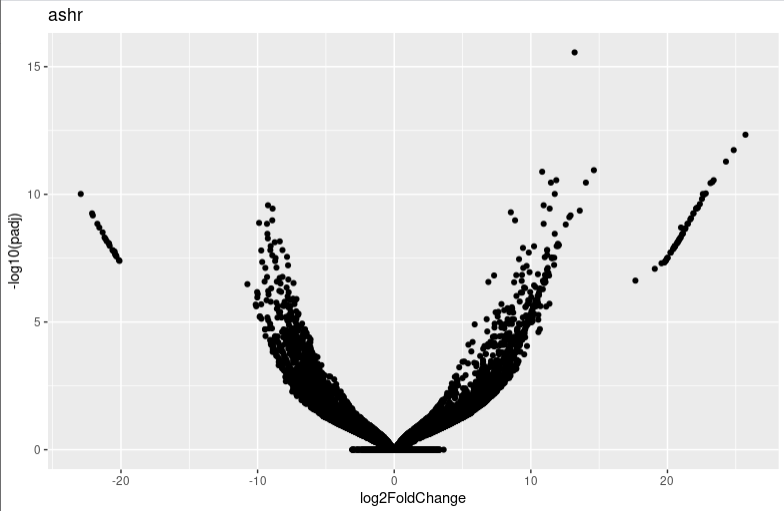
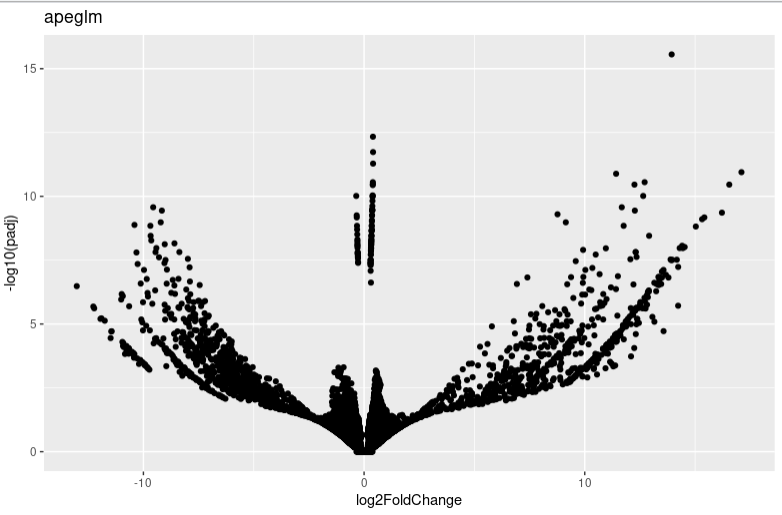
Here is the code, and rawdata
https://github.com/shangguandong1996/picture_link/blob/main/WFX_count_Rmatrix.txt
# Prepare -----------------------------------------------------------------
# load up the packages
library(DESeq2)
library(dplyr)
library(ggplot2)
# Set Options
options(stringsAsFactors = F)
# load up the data
data <- read.table("rawdata/count/WFX_count_Rmatrix.txt",
header = TRUE,
row.names = 1)
coldata <- data.frame(row.names = colnames(data),
type = rep(c("Fx593", "Fx600"), each = 2))
# DESeq2 ------------------------------------------------------------------
dds <- DESeqDataSetFromMatrix(countData = data,
colData = coldata,
design= ~ type)
keep <- rowSums(counts(dds)) >= 10
dds <- dds[keep,]
# PCA
vsd <- vst(dds)
plotPCA(vsd, intgroup = "type")
dds <- DESeq(dds)
lfc_plotVolcano <- function(type){
res_lfc <- lfcShrink(dds = dds,
type = type,
coef = "type_Fx600_vs_Fx593")
as_tibble(res_lfc) %>%
mutate(padj = case_when(
is.na(padj) ~ 1,
TRUE ~ padj
)) %>%
ggplot(aes(x = log2FoldChange, y = -log10(padj))) +
geom_point() +
ggtitle(type)
}
lfc_plotVolcano("ashr")
lfc_plotVolcano("apeglm")
Best wishes Guandong Shang

Thanks for your reply, Dr Love.
I just have another question about pre-filtering 0 count genes. Accoring to your manual, it seems that pre-filter low count is not necessary because there is more strict filtering on
resultfunction.But as you can see, in this condition, pre-filtering or not will make a big infulence on the volcano plot shape. I am just curious why?
Best wishes
Guandong Shang
I’m recommending it for you here aside from the point above that it’s not required for null hypothesis pvalue generation and multiple testing.
Hi, Dr love.
I am wondering whether
lfcShrinkfunction will not applyindependent filtering on the mean of normalized countswhileresultfunction will. But if it is true, should I pre-filtering low count gene each time before running DESeq function if I want to get the lfcShrink log2FoldChange result?Best wishes
Guandong Shang
The lfcShrink function produces posterior effect sizes. Those don’t involve independent filtering or multiple testing.