
we have a data set of 48 samples mapped with STARsolo to create a sparse matrix. After reading the mtx file into a SingleCellExperiment object we would like to normalize it by deconvolution using the computeSumFactors function from scran.
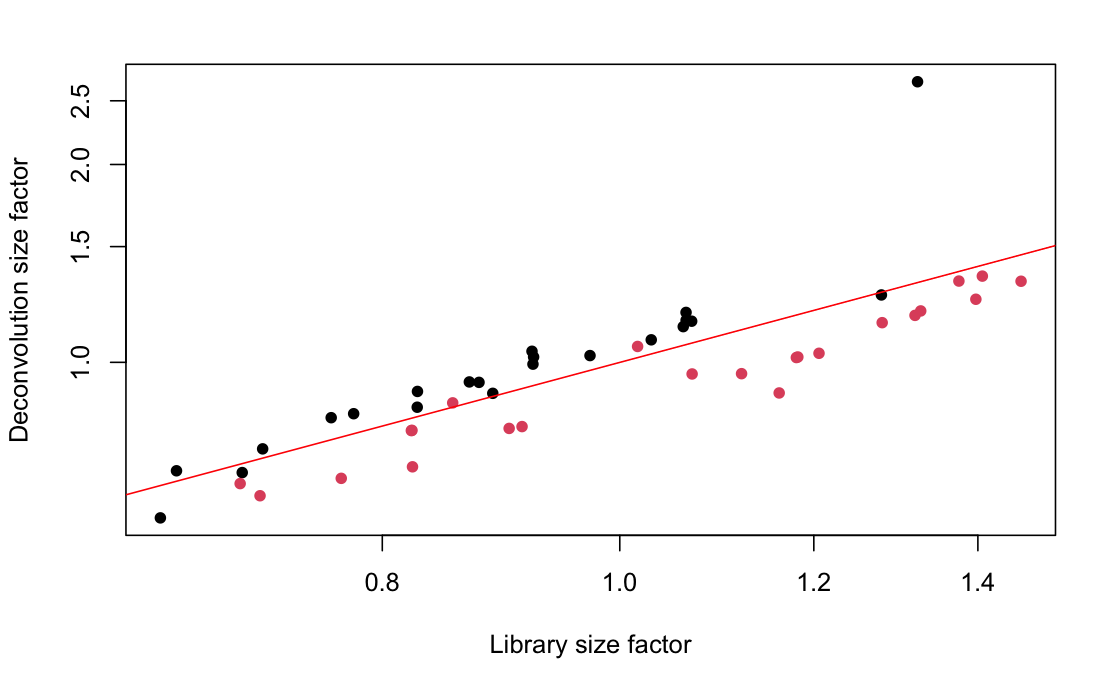
After qc we have only 45 samples left, each represent a separate cell, in two conditions. Interestingly, both the qc as well as the scatter plot of library size show a nice split based on this two conditions (see the two images below). red and black dots are the two conditions in the data set.
A short snippet if the workflow is also attached below.
sce <- SingleCellExperiment(assays = list(counts = cts) )
...
stats <- perCellQCMetrics(sce, subsets=list(Mito=mito))
sce <- sce[,!qc$discard]
lib.sf.sce <- librarySizeFactors(sce) # lib size factors
clust <- quickCluster(sce, min.size = 1) # needed to be reduced for a successful run.
deconv.sf.sce <- calculateSumFactors(sce, cluster=clust) # deconvolution size factors
Is it possible to run a normalization and scaling by deconvolution with such a small data set?
I read here that it might be possible if the scatter plot show a nice correlation of the deconvolution factors and the size factors. But in there, there were three times the amount of cell as we have. In my opinion, they do correlate here quite nicely, but the separation on condition is a bit strange.
I would appreciate the advice.
thanks
Assa
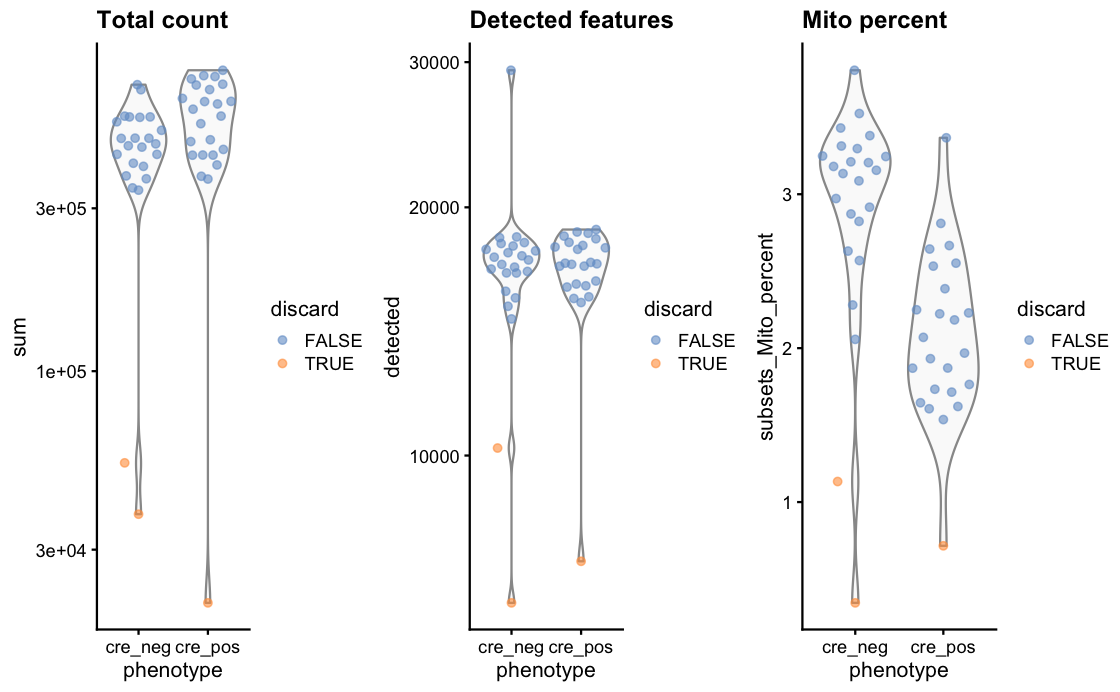
Do I read this correctly that you have about 15k cells detected in almost all of these samples? If so then then I would simply run the default normalization from edgeR (or similar packages) as the point of the deconvolution metjod (pooling cells/samples followed by size factor estimation) is to compensate the abundance of genes with zeros, which here is probably not much of a concern.
edgeR::calcNormFactors()has amethod='TMMwsp'that says it would deal with many zeros, maybe try that as well, despite results are probably similar.no. each sample is one single embryo in this case (so, two cells). I have mapped them with STARsolo from SMART-seq samples. This is the first time I analyzed this kind of data, but if I understand it correctly, the
detected featurescolumns tells me how many genes were found ( as SMART-seq doesn't have UMIs), while thecount totalcolumns shows the number of reads in the samples.Do you still think it make sense to use the
edgeRnormalization method?